Ks. artikkelin pdf-versio «http://www.fimnet.fi/cl/laakarilehti/pdf/2014/SLL42014-219.pdf»1 Lääkärilehden sivuilla (vaatii FiMnet-tunnuksen).
Perinnölliset aineenvaihduntasairaudet ovat laaja joukko sairauksia, joissa yleensä yhden geenin virhe aiheuttaa jonkin entsyymin toiminnan osittaisen vajauksen tai täydellisen puutoksen. Oireet voivat alkaa missä iässä tahansa sikiövaiheesta vanhuuteen asti. Oireet johtuvat entsyymin normaalisti tuottaman ainesosan puutteesta tai substraattina toimivan aineen patoutumisesta ja kertymisestä elimistöön. Suurin osa näistä sairauksista periytyy peittyvästi, joten käytännössä ne vaikuttavat yksittäistapauksilta (1). Perinteisesti aineenvaihduntasairauksia on pidetty lastentauteina, mutta tämä käsitys on muuttumassa.
Yksittäiset metaboliset sairaudet ovat hyvin harvinaisia, mutta joukkona ne ovat yleisiä. Kokonaisilmaantuvuuden uskotaan olevan yli 1:2 000 (2). On todennäköistä, että jokainen lääkäri tapaa jossain vaiheessa uraansa potilaan, jolla on perinnöllinen metabolinen häiriö. Aineenvaihdunnan häiriöitä tunnetaan nykyään yli 500, joista muutama kymmenen voi alkaa oireilla vasta aikuisiässä (3). Britanniassa on pitkät perinteet aikuisten aineenvaihduntasairauksien hoidosta seitsemässä eri keskuksessa (4). Britannian lukuihin suhteutettuna Suomessa saattaa olla perinnöllinen aineenvaihduntasairaus jopa yli 500:lla aikuisella.
Aikuisiällä oireisiksi tulevat aineenvaihduntasairaudet voidaan jakaa patofysiologisesti kolmeen luokkaan.
Ensimmäisen ryhmän sairaudet ovat akuutteja tai kroonisia myrkytyksen kaltaisia oireita aiheuttavat häiriöt, esimerkiksi ureakiertotaudit, hemokromatoosi, porfyria ja homokystinuria. Näiden sairauksien diagnostiikka on melko selkeää ja useimpiin on olemassa hyvä hoito.
Toisena tulevat energiametabolian häiriöt, jotka aiheuttavat energiantuoton tai -käytön häiriön maksassa, lihaksissa, sydämessä, aivoissa tai muissa kudoksissa. Tällaisia tauteja ovat esimerkiksi mitokondriotaudit ja rasvahappo-oksidaation häiriöt. Niiden diagnostiikka on haastavaa ja vain osaan on hoitoa tarjolla.
Kolmantena ovat monimutkaisten molekyylien häiriöt, kuten Fabryn tauti ja Gaucherin taudin tyypit 1 ja 3. Näissä sairauksissa oireet ovat pysyviä ja eteneviä, eivätkä niihin vaikuta ulkoiset tekijät kuten ravinto. Akuuttihoitoa näihin ei ole, mutta yhä useampaan on tarjolla vajaasti toimivan entsyymin korvaushoito estämään lisäkomplikaatioita.
Synnynnäistä aineenvaihduntasairautta potevien lapsipotilaiden hoito on Suomessa keskitetty ensisijaisesti yliopistosairaaloihin. Hoitoa tukee tutkimuksen kautta kehittynyt erityisosaaminen, esimerkiksi lysinurinen proteiini-intoleranssi (LPI) -potilaiden keskus TYKS:ssä sekä mitokondriaalisten sairauksien tutkimus Helsingin ja Oulun yliopistoissa.
Aikuispotilaiden seuranta on lähes täysin joko lastentautiyksiköiden tai edellä mainittujen erityisosaamiskeskusten vastuulla, tai potilaat ovat hajaantuneet yksittäispotilaiksi eri erikoisaloille. Suomessa ei ole aikuismetabolisia keskuksia eikä juurikaan näihin sairauksiin perehtyneitä lääkäreitä, joiden seurantaan voisi aikuistuvia potilaita siirtää jatkohoitoon. Aiemmin on arvioitu, että lapsipotilaista vain noin 11 % selviytyy aikuisikään (4), mutta tehostuvan diagnostiikan ja uusien hoitojen ansiosta aikuisikään selviytyvien potilaiden osuus todennäköisesti kasvaa.
Monessa länsimaassa vastasyntyneiden aineenvaihduntatauteja seulotaan tandemmassaspektrometrialla, minkä avulla tapauksia löydetään aiempaa varhaisemmin. Tämä saattaa johtaa uusien tapausten löytymiseen myös aikuisikäisiltä perheenjäseniltä ja lähisukulaisilta. Tämänkaltaisen vastasyntyneiden seulonnan valtakunnallisesta käynnistämisestä myös Suomessa käydään parhaillaan vilkasta keskustelua.
Nuoruus- tai aikuisiässä oireisiksi tulevat perinnölliset aineenvaihduntasairaudet voivat jäädä löytymättä ja hoitamatta, elleivät aikuisia hoitavat lääkärit tunnista tilanteita, joissa näitä sairauksia tulisi epäillä. Tilannetta hankaloittaa myös aikuisten aineenvaihduntasairauksiin perehtyneiden lääkärien ja siten konsultaatiomahdollisuuksien vähäisyys.
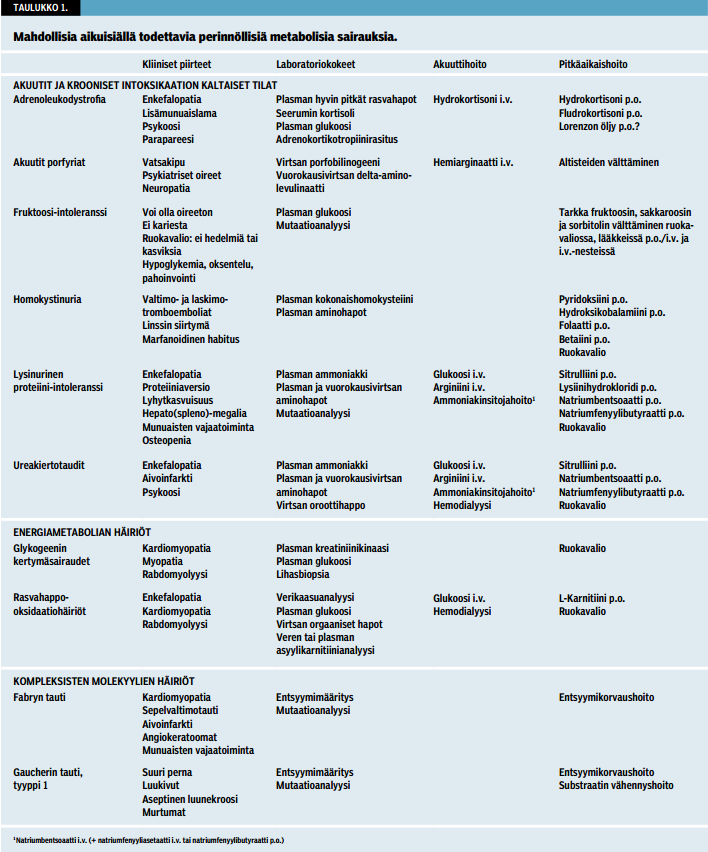
Aikuisiällä oireiseksi tulevat perinnölliset aineenvaihduntasairaudet (taulukko «»1) manifestoituvat usein psykiatrisin tai neurologisin ilmentymin ja oirein (3). Näitä voivat olla esimerkiksi atyyppinen psykoosi, masennus, epäselvä tajuttomuus, perifeerinen neuropatia, pikkuaivoataksia, spastinen parapareesi, dementia, liikehäiriö tai epilepsia.
Perinnöllisen aineenvaihduntasairauden merkki voi olla statuslöydösten ja oireiden vaihtelu, etenkin jos ne aiheutuvat tai pahenevat paastosta, liikunnasta, kuumeesta, synnytyksestä tai muusta katabolisesta tilasta.
Aikuisen oireet ovat voineet olla lieviä ja epämääräisiä tai niitä on esiintynyt useassa eri elinjärjestelmässä, jotka kuuluvat eri erikoisalojen piiriin. Taudin kaikkien ilmentymien kehittyminen voi kestää kymmeniä vuosia, jolloin kliinikko ei osaa epäillä sairautta sen ensimmäisten oireiden alkaessa. Diagnoosi voi pahimmillaan viivästyä jopa vuosikymmeniä.
Toisinaan aikuispotilaan ensimmäiset kliiniset merkit sairaudesta ovat olleet esillä jo lapsena, mutta diagnoosiin ei ole vielä silloin päästy tai epäilys ei ole herännyt, koska koko sairautta tai sen lieväoireista muotoa ei ehkä ollut vielä edes kuvattu oppikirjoissa tai julkaisuissa. Hyvä anamneesi voi paljastaa esimerkiksi kasvun hidastuman vauvana tai lapsena, tai silmän linssin tai sarveiskalvon samentuman nuorella iällä. Potilaalla voi olla useita toisiinsa huonosti liittyviä eri elinjärjestelmien häiriöitä ja yhden tai usean eri neurologisen alueen yhtäaikainen löydös, esimerkiksi pikkuaivoataksia ja perifeerinen neuropatia, sekä tuntemattomasta syystä suurentunut perna tai poikkeava iho- tai silmänpohjalöydös.
Kliiniset tilanteet, joita lääkäri voi kohdata aikuispotilaan sairastaessa tietämättään aineenvaihduntasairautta, voidaan jakaa karkeasti kolmeen ryhmään. Ensimmäisenä ovat akuutit toistuvat oireet, kuten tajuttomuus, ataksia, oksentelu, metabolinen asidoosi, rasituksen siedon heikkous, sydämen, munuaisten, maksan tai muun elimen vajaatoiminta. Toisena ryhmänä ovat krooniset etenevät neurologiset oireet, esim. epilepsia, neurologinen "rapistuminen" ja psykiatriset oireet. Kolmantena ovat potilaat, jotka ovat lapsuudesta lähtien välttäneet tiettyjä ruoka-aineita. Esimerkiksi ureakiertotautia sairastavat välttävät proteiinia ja fruktoosi-intoleranssia sairastavat hedelmien syöntiä.
Aineenvaihduntasairauksien seulonnassa voidaan käyttää yksinkertaisia laboratoriokokeita, jotka voivat paljastaa metabolisen häiriön. On tärkeä muistaa sekä tehdä sopivia seulontatestejä, esim. mitata plasman ammoniakkipitoisuus, että osata tulkita tavanomaisia kokeita, kuten glukoosia ja verikaasuanalyysiä, pitäen mielessä myös aineenvaihduntasairaudet. Perusterveydenhuollon päivystyspisteet keskittyvät usein laadukkaan laboratoriopalvelun yhteyteen, jolloin näitä tutkimuksia on saatavilla.
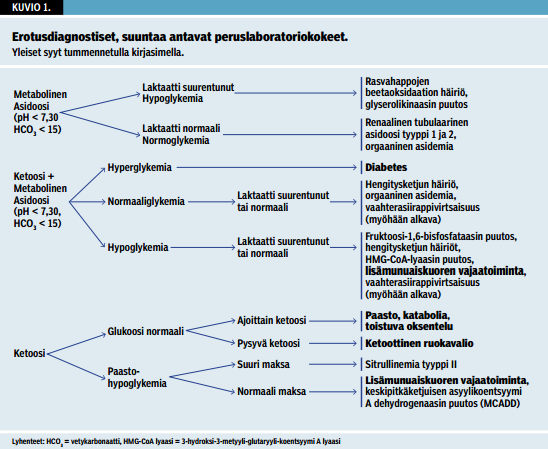
Hypoglykemia yhdessä asidoosin kanssa viittaa metabolisen häiriön mahdollisuuteen. Plasman ammoniakkipitoisuus, veren tai plasman laktaatti ja virtsan tai veren ketoaineet ovat perustutkimuksia, joiden avulla voidaan päästä metabolisen häiriön jäljille (kuvio 1 «»2) (1).
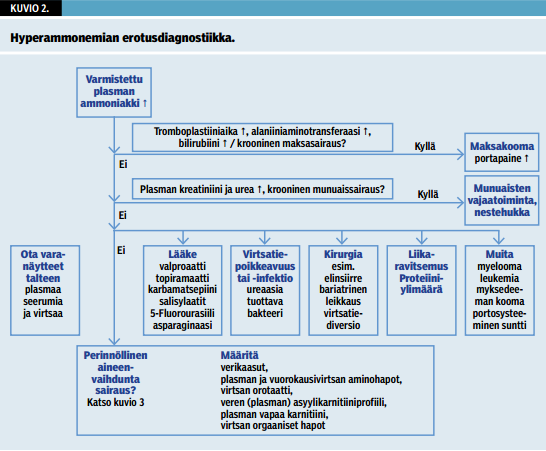
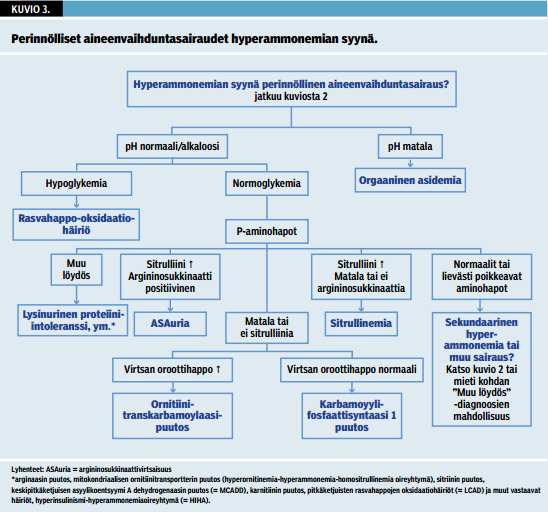
Plasman ammoniakkipitoisuus tulisi määrittää kaikilta tajuttomilta potilailta. Aikuiselle hyperammonemian voivat aiheuttaa useat muutkin erotusdiagnostiikassa huomioon otettavat tilat (kuvio 2 «»3) kuin synnynnäinen aineenvaihduntasairaus (kuvio 3 «»4). On silti tärkeää muistaa, että joskus kuviossa 2 «»3 esitetyt tilat voivat tuoda esiin piilevän aineenvaihduntasairauden (1).
Mikäli seulontaluonteiset laboratoriotulokset tai taudinkuva viittaavat perinnölliseen aineenvaihduntasairauteen, voidaan seuraavaksi tutkia suunnatusti plasman ja keräysvirtsan aminohapot, virtsan orotaatti, asyylikarnitiinit kuivapisaranäytteestä ja virtsan orgaaniset hapot. Näitä tutkimuksia ei yleensä saa päivystysaikana, minkä vuoksi tulee ottaa talteen varanäytteet seerumia, plasmaa ja virtsaa. Niistä voidaan tarvittavalla kiireellisyydellä määrittää aineenvaihdunnan dekompensaatiovaiheessa diagnostisesti poikkeavina esiintyviä metaboliitteja.
Edellä mainituilla tutkimuksilla voidaan diagnosoida yli 30 erilaista metabolista häiriötä. Akuutissa epäilyttävässä tilanteessa on tärkeää ottaa virtsan ja plasman varanäytteet talteen pakastimeen, jolloin myöhemmin voidaan tarvittaessa tehdä lisätutkimuksia. Näiden muistamisen helpottamiseksi on useissa keskuksissa (etenkin lastentautiyksiköissä) luotu aineenvaihduntanäytteiden tutkimuspaketti, johon varanäytteet sisältyvät. Tällainen tutkimuspaketti tulisi kehittää myös aikuisia hoitavissa yksiköissä. Useissa sairauksissa diagnoosiin voi päästä taudinkuvan perusteella valituilla kohdennetuilla laboratoriokokeilla, entsyymiaktiivisuuden määrityksillä tai geenitutkimuksilla.
Aikuispotilailla tajunnanhäiriön taustalla on useimmiten sekundaarinen syy, kuten infektio tai myrkytys. Useimmilta tajuttomilta potilailta tutkitaan verikaasuanalyysi ja veren glukoosipitoisuus. Asidoosin ja hypoglykemian yhdistelmää ei yleensä mietitä sen pidemmälle, vaan molemmat hoidetaan oireenmukaisesti. On hyvä muistaa, että tajuttoman potilaan asidoosin ja hypoglykemian taustalla voi olla primaarinen aineenvaihduntahäiriö, kuten orgaaninen asidemia.
Lievä hyperammonemia, joka ei aiheuta tajuttomuutta, voi ilmetä psykiatrisina oireina. Potilaalla, jolla on akuutit psykiatriset oireet yhdessä alentuneen tajunnantason kanssa, tulisi epäillä metabolista intoksikaatiota. Oireet voivat olla ajoittaisia ja liittyä katabolisiin tilanteisiin.
Ureakierron entsyymien vaikeat toiminnanvajaukset todetaan yleensä jo varhain lapsuudessa (5). Ensimmäinen kohtaus johtaa hoitamattomana kuolemaan ja hoidettunakin voi aiheuttaa eriasteisen aivovaurion. Mikäli entsyymin toiminnanvajaus on vähäisempi, oireita voi esiintyä lapsuudessa lievinä tai ohimenevinä, tai sitten niitä ei ole lainkaan. Osa potilaista on sopeuttanut ruokavalionsa niin, ettei saa oireita. Anamnestinen tieto ravinnon proteiinin huomattavasta omatoimisesta rajoittamisesta lapsuudesta lähtien voi viitata proteiini-intoleranssiin ja proteiinin aiheuttamaan plasman ammoniakkitason nousuun.
Aikuisella tyypillisin hyperammonemian oire on "intoksikaatio" elimistön stressitilan yhteydessä plasman ammoniakkipitoisuuden suurentuessa (taulukko «»1). Tyypillisiä stressitilanteita, joiden jälkeen ammoniakkipitoisuus suurenee, ovat esimerkiksi synnytyksen jälkivaihe, leikkaukset ja vaikeat traumat sekä kuumetaudit tai toisaalta äkillinen, suuri proteiinimäärä ruokavaliossa tai paastoaminen. Aiemmin "terve" henkilö voi muuttua sekavaksi ja käyttäytyä poikkeavasti. Hänen tajuntansa taso voi laskea nopeastikin ja johtaa epäselvään tajuttomuuteen. Ajoittain hänellä voi olla pahoinvointia, oksentelua ja päänsärkyä. Neurologisina oireina voi olla myös hemiplegia (ns. metabolinen aivoinfarkti), progressiivinen spastinen tetraplegia tai kouristuksia. Akuuttitilanteen jälkeen voi asianomaiselta itseltään tai omaisilta saatu anamneesi paljastaa ruokavalion spontaanin rajoittumisen, äkkikuolemia lähisuvussa, kehityshäiriön tai huonosti edistyneen kasvun lapsuudessa.
Eräässä tapauksessa aiemmin terveen 30-vuotiaan naisen sekavuutta ja tajuttomuutta epäiltiin synnytyksenjälkeiseksi psykoosiksi ennen plasman ammoniakin määritystä, joka oli 500 mmol/l (normaali < 50 mmol/l). Kyseessä oli aiemmin diagnosoimaton ureakiertotauti, karbamoyylifosfaattisyntetaasi 1:n puutos (6).
Ureakiertotautipotilaan alkudiagnoosin tulisi tapahtua tehokkaasti, koska seerumin suuri ammoniakkipitoisuus on neurotoksinen ja voi vahingoittaa aivoja pysyvästi. Nopea ammoniakkipitoisuuden määritys pätevässä laboratoriossa on tärkeää. Ammoniakkia sitova ja poistava hoito tulisi aloittaa heti epäiltäessä ureakiertotautia. Potilas tulee siirtää teho-osastolle ja aloittaa hoitokaavion mukainen hoito, jonka ohjeet löytyvät parhaiten päivitettynä sivustolta www.bimdg.org.uk. Samanaikaisesti käynnistetään etiologiset selvitykset.
Metabolinen aivoinfarkti on hyvin harvinainen löydös, joka voi liittyä mm. mitokondriotauteihin, orgaaniseen asidemiaan, ureakiertotauteihin tai Fabryn tautiin. Pään kuvantamistutkimuksista voi saada vihjeen edetä diagnostiikassa oikeaan suuntaan.
Myös homokystinuriassa (taulukko «»1) aivolaskimoiden tai -valtimoiden tromboemboliat tai valtimodissekaatio voivat aiheuttaa neurologisia puutoksia (2,3). Tauti lisää veren hyytymisalttiutta, jonka lisäksi se voi aiheuttaa silmän linssin siirtymän (iridodoneesilöydös, kts. YouTube), vaikean myopian, skolioosin, osteoporoosin ja häiritä keskushermoston kehitystä. Ainoana löydöksenä voi olla selittämätön valtimotukos tai tyypillisimmin raajan syvä laskimotukos tai keuhkoveritulppa. Tanskassa, jossa on 5,5 miljoonaa asukasta, on kahdessa sairaalassa löydetty homokystinuria 10:ltä nuorelta aikuiselta (7). Marfanoidiset piirteet liittyvät vaikeisiin tapauksiin.
Suuntaamattomalla laskimotukospotilaiden plasman homokysteiinin määrityksellä löytyy suurentunut pitoisuus vain noin yhdeltä 2 500 potilaasta. Määritys kannattanee tehdä potilaalle, jolla on toistuvia laskimotukoksia tai sekä laskimo- että valtimotukos ilman selvää etiologista tekijää ja niille, joilla on muita homokysteinurian kliinisiä löydöksiä, kuten linssin siirtymä, vaikea myopia, osteoporoosi, skolioosi tai kehityshäiriö. Plasman kokonaishomokysteiini on selvissä tapauksissa yli 100 mmol/l (normaali < 15 mmol/l). Homokysteiinin määritys ei ole luotettava tutkimus osana plasman aminohappotutkimusta, koska se mittaa vain homokystiiniä, eikä kokonaishomokysteiiniä. Homokysteiinipitoisuutta suurentavat myös B12-vitamiinin tai folaatin puute, vaikea munuaisten vajaatoiminta ja lääkkeistä metotreksaatti, trimetopriini, fenytoiini, sulfasalatsiini ja niasiini. Plasman aminohappomäärityksessä todettava suuri metioniinipitoisuus sopii homokystinuriaan. Metioniinipitoisuus on normaali sekundaarisissa hyperhomokysteinemioissa.
Homokystinurian hoito voi olla helppoa ja hoidon tulokset erinomaisia pyridoksiinille (B6-vitamiini) reagoivassa tautimuodossa (2). Pyridoksiinille reagoimattoman tautimuodon hoito puolestaan voi olla hyvin haastavaa.
Esimerkkitapauksessa 62-vuotias nainen oli tutkimuksissa multippelien aivoinfarktien takia. Hänellä oli ollut syvä laskimotukos 58-vuotiaana, osteoporoosi oli todettu 56-vuotiaana ja molempien silmien linssin siirtymä 20-vuotiaana. Plasman kokonaishomokysteiinipitoisuus pieneni vitamiinihoidoilla (405 mmol/l:sta 50 mmol/l:aan). Toisessa tapauksessa vitamiinihoidon ansiosta potilaalle ei kehittynyt komplikaatioita 56 ikävuoteen mennessä, koska sairaus diagnosoitiin sukulaistapauksen vuoksi 14-vuotiaana, jolloin aloitettiin vitamiinihoidot.
Aiemmin terveelle aikuiselle liikunnan aiheuttama rabdomyolyysi johtuu usein metabolisesta lihassairaudesta (taulukko «»1). Jos rabdomyolyysi toistuu tai jos ensimmäisenkin rabdomyolyysin lisäksi anamneesissa on liikunnan aiheuttamaa lihaskipua tai -kramppeja, on noin joka viidennellä potilaalla sen taustalla todennäköisesti perinnöllinen mitokondriaalinen rasvahappo-oksidaation häiriö (8). Akuutin vaiheen aikana tehty asyylikarnitiinianalyysi kuivapisaranäytteestä voi paljastaa taustalla olevan häiriön. Myöhemmin diagnoosin teko on erityisen haastavaa. Myös glykogeenin kertymäsairaudet (tyyppi V ja VII) voivat aiheuttaa rabdomyolyysin (2).
Lihaskipu voi liittyä rasitukseen joko varhaisvaiheessa tai vasta pitkässä rasituksessa riippuen siitä, missä energia-aineenvaihdunnan osassa häiriö on.
Esimerkkitapauksessa 35-vuotiaalla naisella oli ollut kahdesti lihaskramppi juostessa. Maratonilla hän alkoi voida huonosti (16 km:n kohdalla) ja sai lihaskramppeja (27 km:n kohdalla), mutta pääsi maaliin. Seuraavana yönä lihakset kramppailivat ja virtsa oli tummaa. Aamulla sairaalassa seerumin kreatiniinipitoisuus oli 256 mmol/l (normaali 50-90 mmol/l) ja kreatiniinikinaasipitoisuus 149 573 U/l (normaali 35-210 U/l). Dialyysihoidolla munuaiset toipuivat. Rasvahappojen beetaoksidaation häiriö (karnitiinipalmitoyylitransferaasi II:n puutos) varmistui geenitutkimuksen avulla.
Aikuisille metaboliset häiriöt aiheuttavat tyypillisesti hypertrofisen kardiomyopatian. Se voi johtua lihassolun energia-aineenvaihdunnan häiriöstä tai makromolekyylien kertymisestä (esim. Fabryn tauti, glykogeenikertymäsairaudet ja pitkäketjuisten rasvahappojen oksidaatiohäiriöt) (taulukko «»1). Usein sydämen johtumisjärjestelmä häiriytyy, ja oireena voi olla rytmihäiriöitä tai äkkikuolema.
Esimerkkitapauksessa 40-vuotiaalle urheilijamiehelle kehittyi maratonin jälkeen vaikea kammiotakykardiataipumus ja hänellä todettiin sydämen kärjen ja väliseinän hypertrofiaa. Selvityksissä sydänlihasbiopsiassa todettiin glykogeenikertymiä viitaten glykogeenin kertymäsairauteen. Sairauden tarkempaa etiologiaa selvitetään.
Onko potilaasi valittanut haisevansa pahalle ilman syytä? Hän voi tuntea hajun itsekin, mutta osa potilaista ei havaitse sitä. Hajun takia hän on voinut eristäytyä muista ihmisistä, käydä psykiatrin vastaanotolla tai häneltä on voitu poistaa nielurisat.
Kyseessä voi olla sekundaarinen tai harvoin primaarinen trimetyyliaminuria eli kalanhajuoireyhtymä (taulukko «»1) (9). Hajua voidaan kuvata kalan, mädän kalan, viemärin, ulosteiden yms. kaltaiseksi. Eräässä tapauksessa potilaan neljävuotias poika ei suostunut menemään äitinsä lähelle makeana kokemansa hajun aikana. Epämiellyttävä haju voi ilmestyä jo lapsuudessa tai vasta 20 ikävuoden jälkeen. Haju voi olla ajoittaista liittyen esimerkiksi kuukautiskiertoon, hikoiluun tai runsaaseen trimetyyliamiinin esiasteiden (esimerkiksi kala) ja koliinin (esimerkiksi kananmuna) saantiin ruoasta.
Diagnoosia varten trimetyyliamiinin ja trimetyyliamiinioksidin pitoisuuksien suhde voidaan määrittää virtsanäytteestä. Hoitoina voidaan käyttää ruokavaliota, johon liitetään tarvittaessa vitamiinikorvaushoito, suoliflooraan vaikuttavia lyhyitä antibioottikuureja, joihin liitetään probiootteja suoliflooran muuntamiseksi, riboflaviinia, sopivia ihonpuhdistusaineita ja mahdollisesti lääkehiiltä.
Jos potilaan tila sopii kliinisesti perinnölliseen metaboliseen häiriöön, kliinikon tulisi ottaa se huomioon erotusdiagnostiikassa yleisempien sairauksien ohessa. Viimeistään silloin, kun potilaan oireet jatkuvat edelleen tai niitä ei ole selvitetty, vaikka yleisten sairauksien normaalit tutkimukset on tehty ja hoidot aloitettu, tulisi mielessä käydä perinnöllisen aineenvaihduntasairauden mahdollisuus. Oire- ja oireyhtymädiagnoosin takana voi olla perinnöllinen metabolinen häiriö - jopa uusi ja aiemmin tunnistamaton tila.
Useat metaboliset häiriöt ovat hoidettavia ja hoito on erittäin palkitsevaa, koska sillä voidaan usein muuttaa taudin kulku tai hoito voi pelastaa hengen. Diagnoosin jälkeen osalle potilaista voidaan tarjota prenataalidiagnostiikkaa, jonka tarkoituksena on joko estää taudin siirtyminen tuleville sukupolville (etenkin X-kromosomiin liittyvissä taudeissa) tai mahdollistaa jälkeläisen sairauden hyvin varhainen hoito.
Neljä esimerkkitapauksista on esitetty kollegoiden Robin Lachmann ja Elaine Murphy luvalla (University College London -sairaala, Arthur Dentin aikuismetabolinen yksikkö).
Inborn errors of metabolism (IEM) are genetic diseases of any metabolic route, and they can present at any age from birth to late adulthood. Paediatricians have traditionally diagnosed and treated the severe forms of these diseases. The milder or late-onset forms of IEMs usually commence or become recognisable in early adulthood; however, they may also appear in late adulthood. Doctors treating adult patients should be aware of typical presentations and the initial laboratory tests that give clues to the presence of a possible IEM. A significant number of IEMs are treatable, and treatment may dramatically alter the course and prognosis of an IEM.