Ks. artikkelin pdf-versio «http://www.fimnet.fi/cl/laakarilehti/pdf/2016/SLL122016-901.pdf»1 Lääkärilehden sivuilla (vaatii FiMnet-tunnuksen).
Perinnölliset hemoglobiinipoikkeavuudet, kuten talassemia ja sirppisoluanemia, ovat yleisiä Kaakkois-Aasiassa ja päiväntasaajan alueen Afrikassa. On arvioitu, että näiden alueiden vastasyntyneistä 5-20 %:lla on perinnöllinen hemoglobiinipoikkeavuus.
Alfatalassemioiden suurin esiintyvyys on Kaakkois-Aasiassa ja Länsi-Afrikassa. Beetatalassemioita löytyy laajana vyöhykkeenä Välimeren ympäristöstä ulottuen Arabian niemimaahan, Intiaan ja edelleen Kaakkois-Aasiaan. Sirppisoluanemiaa esiintyy eniten Afrikassa päiväntasaajan alueella.
Suomessa asui vuonna 2013 noin 100 000 henkilöä, jotka olivat lähtöisin perinnöllisen hemoglobiinipoikkeavuuden yleisiltä esiintymisalueilta (1). Tuoreiden ennusteiden mukaan tämä luku tulee kasvamaan vielä 30 000-50 000:lla vuoden 2016 loppuun mennessä. Varovaisestikin arvioiden Suomessa tulee olemaan useita satoja, mahdollisesti jopa tuhat, periytyvän hemoglobiinipoikkeavuutensa vuoksi tietoa, seurantaa ja hoitoa tarvitsevaa potilasta.
Hemoglobiinipoikkeavuuksista kärsivät maahanmuuttajataustaiset henkilöt ovat hyvin epäyhtenäinen joukko lapsia, nuoria ja aikuisia. He ovat Suomessa tai ulkomailla syntyneitä ja heidän koulutustaustansa on hyvin kirjava (ks. esimerkkipotilaat, liite 1. www.laakarilehti.fi > Sisällysluettelot «http://www.fimnet.fi/cl/laakarilehti/pdf/2016/SLL122016-901.pdf»1).
Monen ulkomailla syntyneen hemoglobiinipoikkeavuus on diagnosoitu jo synnyinmaassa. Kehittyvistä maista tulevilla ei yleensä ole ollut mahdollisuutta saada komplikaatioita ehkäisevää hoitoa tautiinsa. Potilaan pitkäaikaisselviytymisen kannalta on olennaista, missä iässä hän tulee kehittyneen terveydenhuollon piiriin ja kuinka hyvin terveydenhoitojärjestelmässä tunnistetaan potilaan ongelmat ja työskennellään komplikaatioiden ehkäisemiseksi.
Tämän katsausartikkelin tavoitteena on parantaa erityisesti perusterveydenhuollossa toimivien terveydenhuollon ammattilaisten kykyä löytää ne potilaat, jotka hyötyvät hemoglobiinipoikkeavuuden diagnosoinnista ja monialaisesta hoidosta.
Talassemia jaetaan kliinisen vaikeusasteen mukaan kolmeen ryhmään: vaikeaan ja keskivaikeaan talassemiaan sekä talassemian kantajuuteen. Talassemian syynä on tavallisimmin peittyvästi periytyvä geenivirhe joko globiinia koodaavissa HBA- tai HBB-geeneissä. Muutos molemmissa geeneissä muokkaa taudinkuvaa. Talassemian komplikaatiot liityvät anemiaan, hemolyysiin ja rautakertymään (2,3).
Vaikean beetatalassemian oireet alkavat pian syntymän jälkeen. Veren hemoglobiinipitoisuus pienenee ja lapsen kasvu pysähtyy. Potilaan perna suurenee, ja jos hän ei saa hoitoa, ilmaantuu noin yhden vuoden iästä lähtien luukystia ja poikkeavat kasvonpiirteet. Hoidoksi potilas tarvitsee säännöllisiä punasolusiirtoja ja rautakuorman purkua lääkkeillä. Rautakelaatio aloitetaan viimeistään noin vuoden kestäneiden säännöllisten punasolusiirtojen jälkeen tai jos plasman suurentunut ferritiinipitoisuus tai magneettikuvaukseen perustuva kudosraudan arvio viittaavat rautakertymään (2). Vaikean talassemian ainoa parantava hoito on allogeeninen kantasolusiirto, jota harkitaan, jos potilaalle on tarjolla sopiva luovuttaja eikä kudosraudan kertymisestä ole ehtinyt syntyä merkittäviä elinvaurioita (4,5).
Keskivaikean talassemian hoidon kulmakivenä on tunnistaa potilaat, jotka tarvitsevat punasolusiirtoja tai rautakelaatiohoitoa normaalin kasvun ja toiminnan takaamiseksi. Osa keskivaikeaa talassemiaa, esim. HbE-beetatalassemiaa tai HbH-tautia, sairastavista potilaista tulee punasolusiirroista riippuvaiseksi lapsuudessa, osa vasta aikuisiällä, osa ei koskaan. Punasolusiirtoja voidaan tarvita myös satunnaisesti lyhyinä jaksoina erilaisissa stressitilanteissa, kuten vaikean sairauden aikana. Punasolusiirroista ja perustaudista kertyvä rautalasti heikentää erityisesti sydämen ja hormoneja tuottavien elinten toimintaa (2,3). Verenkierrossa vapaana liikkuva hemi vaurioittaa edelleen suonten endoteeliä ja altistaa keskivaikeaa talassemiaa sairastavat potilaat trombooseille.
Sekä vaikeaa että keskivaikeaa talassemiaa sairastavat potilaat tarvitsevat säännöllistä foolihappolisää ja ohjeet vähän rautaa sisältävästä ruokavaliosta.
Talassemian kantajat ovat yleensä oireettomia eivätkä tarvitse rutiinimaista seurantaa, erityishoitoja tai -tutkimuksia. Varmistamalla diagnoosi kantajat välttyvät kuitenkin tarpeettomalta rautalääkitykseltä ja heidät voidaan ohjata perinnöllisyysneuvontaan.
Suurin osa sirppisoluanemioista aiheutuu homotsygoottisesta geenivirheestä globiinia koodaavassa HBB-geenissä (HbSS), joka altistaa hemoglobiinimolekyylit palautumattomalle muodonmuutokselle eli sirppiytymiselle. Sirppiytyneet punasolut liimautuvat sekä toisiinsa että suonen seinämiin, mikä johtaa suonten tukkeutumiseen ja punasolujen hajoamiseen. Ilmiön muuttumista akuutiksi kutsutaan sirppisolukriisiksi. Kriisit aiheuttavat kudoksiin happivajausta, infarkteja ja potilaalle kovia kipuja.
Heterotsygoottiset HbAS-potilaat eli sirppisoluanemian kantajat ovat yleensä oireettomia eivätkä tarvitse tämänhetkisen tiedon mukaan säännöllistä seurantaa. Ääritilanne, kuten vaikea sepsis, saattaa kuitenkin laukaista myös taudin kantajien punasolujen sirppiytymisen. Sirppikriisejä, jotka useimmiten ovat vaikeusasteeltaan lievempiä, ilmenee myös potilailla, jotka sairastavat hemoglobiinipoikkeavuuksien yhdistelmiä, kuten HbSC:tä, HbS-alfatalassemiaa ja HbSD:tä. Edellä mainituista poiketen HbS-beetatalassemia aiheuttaa vaikeaoireisen sirppiytymistaudin.
Potilaiden ensimmäiset kriisit saattavat ilmetä jo alle vuoden iässä ja muut oireet ensimmäisen parinkymmenen ikävuoden aikana. Infektio- ja pernasekvestraatioriskin (hypovolemiaan johtava pernan sirppikriisi) vuoksi pienten lasten sirppisoluanemian diagnoosilla on kiire. Pernan toiminnan hiipumisen ja sepsisriskin vuoksi imeväisille tulee aloittaa heti diagnoosin varmistuttua penisilliini.
Kylmän, hapenpuutteen (vuoristo, lentomatkat, nukutus) ja voimakkaan ruumiillisen rasituksen sekä kuivumisen välttäminen vähentävät sirppikriisejä. Oireileville potilaille hematologi voi aloittaa estolääkityksen hydroksiurealla. Jos oireet eivät helpotu tai ilmaantuu aivoverenkierron häiriöitä, tarvitaan punasolusiirtohoitoa (6,7). Siirtohoito ei ole ongelmatonta, vaan johtaa ilman kelaatiota rautalastin kertymiseen ja mahdollisiin elinvaurioihin. Sirppisolupotilailla on myös merkittävä punasoluimmunisaatioriski sekä riski sairastua henkeä uhkaaviin infektioihin (6,7). Kaikki sirppisoluanemiapotilaat tarvitsevat myös pneumokokkirokotesuojan.
Punasolujen jatkuva hiljainen sirppiytyminen ja sitä seuraava krooninen hapenpuute vaurioittavat elimistöä (mm. aivoja, keuhkoja, luuydintä, pernaa, verkkokalvoa, munuaistubuluksia, sydäntä ja luita) ilman oireisia kriisejäkin. Useimmat sirppisoluanemian krooniset elinvauriot ilmaantuvat varhaisella aikuisiällä, 20-30-vuotiaana, ja lyhentävät elinikää (8).
Erityisen pelätty komplikaatio on aivoverisuonten vaurioituminen, joka altistaa oireisille aivoinfarkteille, oppimisvaikeuksille ja sosiaalisille ongelmille (9). Jopa joka kymmenes potilas saa aivoinfarktin ennen 20. ikävuottaan. Monille kehittyy myös Moya-Moya-oireyhtymä tai uusia pienten kollateraalisuonten verkostoja sisemmän kaulavaltimotukoksen vuoksi. Oppimisvaikeudet voivat pahentua kouluvuosien aikana ja vaativat seurantaa.
Aivoverenkiertohäiriöiden ehkäiseminen on kansantaloudellisesti järkevää. Tämänhetkisen tiedon mukaan tehokkain, joskaan ei riskitön, keino ovat säännölliset punasolusiirrot. Niillä pyritään sirppiytyvien solujen osuuden vähentämiseen ja hapenkuljetuksen parantamiseen aivoissa (9,10). Neurologin tekemä transkraniaalinen dopplerkaikukuvaus (TCD) pystyy poimimaan tiheämpään seurantaan ne lapset ja nuoret, joilla on erityisen suuri aivoinfarktiriski (9,10). Poikkeava löydös on aihe aloittaa säännölliset punasolusiirrot (10). Kajoamattomana tutkimuksena dopplerkaikukuvaus on edullinen ja turvallinen keino diagnosoida ja seurata verisuonitaudin etenemistä. Tutkimus voidaan toistaa tarvittaessa myös päivystysolosuhteissa. Rutiinimaista TCD-seurantaa suositellaan 2-16-vuotiaille potilaille (10). Sen merkitys aikuispotilaiden seurannassa on toistaiseksi avoin.
Pienten lasten vertamuodostavien kantasolujen siirroista on viime aikoina saatu hyviä hoitotuloksia. Siirtohoitoa tullaan jatkossa tarjoamaan kaikille oireista sirppisoluanemiaa sairastaville lapsipotilaille, joilla on kudossopiva sisarus (5). Sisarusluovuttaja on useimmiten itse taudin kantaja, mikä ei sellaisenaan estä kantasolujen luovutusta. Kantasolusiirrot rekisteriluovuttajalta eivät ole sirppisoluanemian rutiinihoitoa. Niitä on toistaiseksi tehty hyvin vähän, sillä sopivien luovuttajien löytymien afrikkalaista alkuperää oleville potilaille on ollut vaikeaa. Sopivia napaveriyksiköitä on löytynyt hieman helpommin. Rekisteriluovuttajan kantasolusiirron tulokset eivät ole olleet yhtä hyviä kuin sisarusluovuttajan, ja niitä suositellaan tehtäväksi ainoastaan osana kokeellista hoito-ohjelmaa kokeneissa keskuksissa sisarussiirtoja tiukemmilla indikaatioilla. Aikuispotilaille kantasolusiirtoja on maailmalla tehty vähemmän. Aikuisten kantasolusiirtoihin on liittynyt ongelmia, jos potilaalle on ehtinyt kehittyä merkittävä rautalasti tai elinvaurioita.
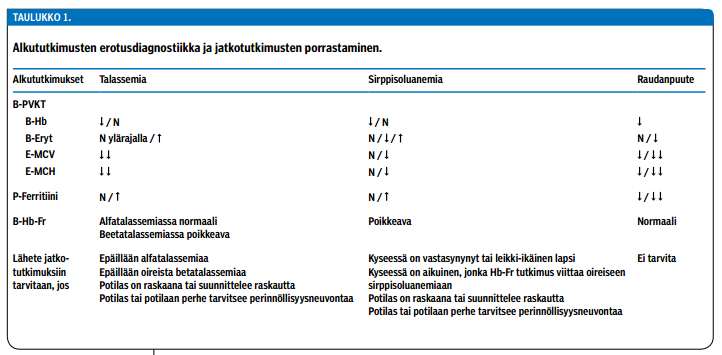
Maahanmuuttajapotilaan ensimmäinen kontakti on usein perusterveydenhuollossa (ks. esimerkkipotilaat). Perusterveydenhuolto vastaa hemoglobiinipoikkeavuuden alkuvaiheen laboratoriotutkimuksista. Hemoglobiinipoikkeavuuden perustutkimuksiin kuuluvat verenkuvan (B-PVKT) lisäksi plasman ferritiinin (P-ferrit) mittaus ja hemoglobiinifraktioiden (B-Hb-Fr) tutkiminen. Riittävät esitiedot ovat oleellinen osa diagnostiikkaa. Löydösten tulkinnassa on tärkeä ottaa huomioon lähiaikojen verensiirrot, mahdolliset samanaikainen raudanpuute- ja megaloblastinen anemia sekä potilaan mahdollinen raskaus. Nämä tiedot on liitettävä myös hemoglobiinifraktiotutkimuksen lähetteeseen.
Talassemiaan (ja HbE-varianttiin) liittyy tyypillinen verenkuva (erytrosytoosi ja voimakas mikrosytoosi), jotka yhdistettynä tietoon potilaan etnisestä syntyperästä viittaavat vahvasti hemoglobiinipoikkeavuuteen. Erotusdiagnostiikassa on otettava huomioon raudanpuuteanemia, jossa punasoluindeksit muistuttavat talassemiaa, mutta punasolumäärä (B-Eryt) ja P-ferrit-pitoisuus ovat pienemmät (taulukko «»1). Talassemiaan ei sovi myöskään se, että E-MCV-arvo on joskus ollut normaali.
Hemoglobiinivarianteissa (hemoglobiinien rakennepoikkeavuuksissa, esim. sirppisolupoikkeavuus) verenkuvalöydökset vaihtelevat suuresti, eikä hemoglobiinipoikkeavuutta voi varmasti diagnosoida verenkuvan perusteella. Hb-varianttien kantajilla (eli heterotsygooteilla) verenkuva on useimmiten normaali tai enintään lievästi poikkeava.
Veren sivelyvalmisteen (B-Morfo) tutkiminen on perinteisesti ollut osa hemoglobiinipoikkeavuuksien selvittelyä. Koska sivelyvalmisteen löydökset jäävät useimmiten epäspesifisiksi, ei tätä tutkimusta tulisi käyttää yksinään perustutkimuksena. Sivelyvalmiste tarjoaa hematologille yhdellä silmäyksellä tiedon potilaan punasolujen koosta, muodosta (sirppisolut) ja polykromasiasta. On muistettava, että perifeerisen veren sivelynäyte kuuluu ottaa potilaasta ennen punasolutankkausta. Myös akuutin hemolyysin yhteydessä (esim. sirppisolukriisi) voi veren sivelyvalmisteesta saada lisätietoa.
B-Hb-Fr-tutkimus sisältää kaikki tarpeelliset proteiinitason tutkimukset, joiden avulla voidaan havaita beetatalassemia ja tavallisimmat hemoglobiinivariantit (HbS, HbE, HbC ja HbD). Alfatalassemiaa ei B-Hb-Fr-tutkimuksella kuitenkaan voida osoittaa. Alfatalassemian diagnostiikka ja kombinoitujen Hb-poikkeavuuksien jatkoselvittelyt onnistuvat ainoastaan geneettisin menetelmin.
Hemoglobiinipoikkeavuuden tarkka diagnostiikka vaatii perustutkimusten lisäksi geenitutkimuksen. Oman kokemuksemme mukaan parhaaseen lopputulokseen päästään tunnistamalla sekä globiinia koodaavien HBA- että HBB-geenien mutaatiot samalla kertaa. Tämä tehdään tutkimuksella, jossa käytetään sekä kyseisten geenien sekvensointia että geenialueiden deleetioiden (häviämien) ja duplikaatioiden (monistumien) osoitusta. Näin toimimalla saadaan selville kombinoidut Hb-poikkeavuudet. Tällä on myös kliinistä merkitystä, sillä esimerkiksi sirppisoluanemiaa sairastavan henkilön mahdollinen alfatalassemian kantajuus vaikuttaa potilaan hydroksiureavasteeseen.
Geenitutkimuksen perusteella on mahdollista antaa perheille perinnöllisyysneuvontaa, jota varten potilaat tulisi ohjata perinnöllisyyslääkärin vastaanotolle. Kantajien tarkalla tunnistamisella ja oireisten lasten varhaisella geenitutkimuksella on merkitystä myös perhesuunnittelun kannalta. Hemoglobiinipoikkeavuuden vaikeusasteesta riippuen osa perheistä voi myös pohtia sikiötutkimuksia tai alkiodiagnostiikkaa. Mikäli tieto mahdollisesta hemoglobiinipoikkeavuudesta tulee esiin alkuraskaudessa, tarvittavat kantajuus- ja mahdolliset sikiötutkimukset voidaan järjestää kiireellisesti konsultoimalla yliopistosairaaloiden sikiötutkimuksiin erikoistunutta yksikköä ja perinnöllisyyslääkäriä.
Hemoglobiinipoikkeavuudet ovat Suomessa ennestään varsin tuntematon tautiryhmä. Väestörakenteen muuttuessa on tärkeää, että koulutusta aiheesta lisätään ja terveydenhoito varautuu tämän potilasryhmän tarpeisiin.
Seurannan järjestäminen vaatii perinteiset toimialarajat ylittävää henkilöstöresursointia ja moniammatillista työryhmää. Seuranta vaatii myös erityistutkimuslaitteita (esim. TCD), sekä työhön motivoituneita ja koulutettuja terveydenhuollon toimijoita. Ranskassa ja Englannissa on käytössä hemoglobiinipoikkeavuuksien osaamiskeskus sekä selkeä työnjako eri toimijoiden välillä (taulukko «»2).
Samanlainen työnjako olisi mahdollista myös Suomessa. Potilaiden alkudiagnostiikka voitaisiin toteuttaa taulukon «»1 mallin mukaisesti. Yliopistosairaalan osaamiskeskus toimisi aktiivisena konsultaatio- ja koulutuspisteenä ja järjestäisi oireisten potilaiden vuosikontrollit erityistutkimuksineen sekä huolehtisi hoito- ja seurantaohjeet kotisairaaloille tai perusterveydenhuoltoon kontrollien väliajaksi (taulukko «»2).
Merkittävä osa hemoglobiinipoikkeavuuksista kärsivistä potilaista elää Suomessa tällä hetkellä ilman heille kuuluvaa terveydenhuollon erityishuomiota. Vaikeutena ovat usein potilaiden ja terveydenhuollon edustajien väliset kulttuuri- ja kielierot. Myös tiedon puute tautikirjoon liittyvistä ongelmista ja siitä, että taudinkulkuun voidaan nykyään vaikuttaa monin tavoin, ovat hoidon ja seurannan esteinä. Väestörakenteen muuttuessa on tärkeää, että koulutusta aiheesta lisätään ja terveydenhoito varautuu tämän potilasryhmän tarpeisiin selkeällä työnjaolla. Tavoitteena on tarjota kaikille hemoglobiinipoikkeavuutta sairastaville potilaille mahdollisuutta asiantuntevaan ja terveystaloudellisesti kestävään seurantaan ja hoitoon.
In Finland in 2013 there were more than 100 000 people originating from countries where haemoglobinopathies, such as thalassaemia and sickle cell disease are common. It has been estimated that by the end of 2016 the number will increase by at least 50%. This means that several hundred patients will be in need of regular health check-ups and treatment due to the haemoglobinopathies.
Neurologic disturbances are the most drastic complications of haemoglobinopathies, especially in patients with sickle cell anaemia. It is known that as many as one in ten (11%) children with sickle cell anaemia suffer an ischaemic stroke before the age of 20. Strokes as well as other complications can, however, be diminished by offering proper routine check-ups and multidisciplinary expertise to the patients at high risk. In order to accomplish this aim, specific diagnostic tools such as Transcranial Doppler (TCD), medical and cell-based treatment and a motivated team of health care professionals are required.
In this article we suggest that primary health care or local hospitals could be responsible for the routine first-line investigations for haemoglobinopathies (blood count, plasma ferritin and Hb isoelectric focusing and cation exchange high performance liquid chromatography) and for picking up the patients at high risk of complications. Genetic testing (HBA- and HBB-gene analyses), more precise screening for risks, specific treatment as well as counselling would then be offered at a university hospital. Despite the increase in the number of people in Finland with haemoglobinopathies, these disorders are still rare, a fact which should be acknowledged when planning optimal care for these patients.