Ks. artikkelin pdf-versio «http://www.fimnet.fi/cl/laakarilehti/pdf/2018/SLL112018-700.pdf»1 Lääkärilehden sivuilla (vaatii FiMnet-tunnuksen).
Trombosytopenia on yleinen löydös lapsilla. Lapsen iästä riippumatta trombosyyttiarvo alle 150 × 109/l katsotaan pienentyneeksi ja syvästä trombosytopeniasta puhutaan, kun trombosyyttiarvo on alle 20-30 × 109/l. Usein lapsen trombosytopenia on suhteellisen vaaraton ja ohimenevä, mutta taustalla voi olla myös jatkoselvittelyjä tai seurantaa vaativa vakava sairaus.
Trombosytopenia voi johtua trombosyyttien lisääntyneestä kulutuksesta, vähentyneestä tuotannosta tai verihiutaleiden siirtymisestä väärään tilaan (sekvestraatio). Ilmeisin sen aiheuttama ongelma on vuototaipumus, joka ilmenee nenäverenvuotona, petekioina ja mustelmina.
Lapsilla yleisin on immunologinen trombosytopenia (ITP). Trombosytopenia voi johtua myös akuutista infektiosta, tai se voi olla merkki perinnöllisestä alttiudesta luuytimen toimintahäiriöön tai oireyhtymästä, jonka muut ilmenemismuodot vaativat seurantaa tai hoitoa.
Idiopaattinen tai immunologinen trombosytopenia (ITP) on autoimmuunitauti, jossa elimistö muodostaa omia trombosyyttejä tuhoavia autovasta-aineita ja trombosyytit vähenevät alle tason 100 × 109/l (1). ITP:n ilmaantuvuus alle 15-vuotiailla on 4,8/100 000 (2). Tyypillinen potilas on alle kouluikäinen lapsi, mutta tautia esiintyy myös vanhemmilla lapsilla ja aikuisilla (2,3). Oireet ilmaantuvat yleensä alle kahdessa viikossa, usein infektion tai rokotuksen jälkeen (4,5). Kliiniseen taudinkuvaan kuuluvat petekiat, purppura, mustelmat ja vuotoalttiuden lisääntyminen muutoin terveellä lapsella (3,6-10). Diagnosointivaiheessa trombosyyttitaso on usein alle 10 × 109/l (3).
Aiemmin immunologinen trombosytopenia luokiteltiin keston mukaan akuuttiin ja krooniseen: akuutti muuttui krooniseksi kestettyään yli 6 kuukautta. Vuonna 2009 luokittelu muuttui: alle 3 kuukautta diagnoosista on uusi ITP, 3-12 kuukautta diagnoosista persistoiva ITP ja taudin kestettyä yli 12 kuukautta krooninen ITP (11). Useimmilla lapsilla trombosyyttitaso on normaali jo 3 kuukauden kuluttua diagnoosista. Lapsipotilaista 15-20 %:lla ITP jatkuu persistoivaksi ja vain 1-2 %:lla se kroonistuu (11,12).
ITP on poissulkudiagnoosi. Tärkeitä tietoja ovat sukuhistoria, oireet, viimeaikaiset infektiot ja rokotukset sekä lääkitys. Huomiota tulee kiinnittää myös dysmorfisiin piirteisiin, jotka saattavat viitata trombosytopeniaoireyhtymiin (taulukko 1 «»1) tai luuytimen perinnöllisiin toimintahäiriöihin. Perusverenkuvan lisäksi tulee tutkia ainakin valkosolujen erittelylaskenta ja perifeerisen veren morfologia. ITP:ssa verihiutaleet ovat usein normaalia kookkaampia, mutta selvästi pienempiä kuin eräille trombosytopeniaoireyhtymille tyypilliset jättitrombosyytit (13). Lisäksi suositeltavaa on tutkia Coombsin koe, virusetiologia (VZV, EBV, parvovirus B19, CMV ja mahdollisesti muitakin viruksia), immunoglobuliinitasot, ANA- ja anti-DNA-vasta-aineet ja hyytymistutkimukset (TT ja APTT). Erotusdiagnostiikassa on hyvä muistaa myös pseudotrombosytopenia, jossa EDTA aiheuttaa verihiutaleiden aggregoitumisen. Trombosyytit voidaan laskea luotettavasti sitraattiputkeen otetusta näytteestä.
Luuydintutkimusta ei ole välttämätöntä tehdä tyypilliselle ITP-potilaalle. Luuydin on tutkittava akuuttivaiheessa vain, jos herää epäily leukemiasta, jolloin potilas on usein selvästi sairas, hänellä todetaan useamman verisolulinjan poikkeavuus ja mahdollisesti lymfadenopatiaa. Luuydintutkimus on tarpeen myös, jos ITP ei reagoi ensilinjan lääkehoitoon (immunoglobuliini-infuusio ja/tai kortikosteroidi) tai se kroonistuu tai seurannan aikana kehittyy muiden verisolulinjojen häiriöitä, jotka voivat viitata laajempaan luuytimen toimintahäiriöön. ITP-potilaan luuydin on morfologisesti normaali, mutta megakaryosyyttien määrä on tyypillisesti selvästi lisääntynyt.
Hoitoa ohjaavat vuoto-oireet, ei trombosyyttien määrä. Jopa lapsilla, joilla on hyvin vähän trombosyyttejä (alle 10 × 109/l), vuodot rajoittuvat yleensä mustelmiin. Toisaalta harvinaisissa vaikeissa keskushermostovuototapauksissa potilailla poikkeuksetta trombosyyttitasot ovat olleet alle 20 × 109/l. Diagnosointivaiheessa vaikea vuoto-ongelma havaitaan noin 3 %:lla lapsista. Vuoto-oireita voidaan arvioida Buchananin ja Adixin asteikolla (14), jossa arvioidaan vuodon yleinen vaikeusaste (0-5) ja erikseen nenäverenvuodon, suun alueen verenvuodon ja ihonalaisen verenvuodon vaikeusaste (0-4) (taulukko 2 «»2).
Vaikeusasteen 1-2 vuotoja pystytään usein hallitsemaan paikallishoidolla, kuten kompressiolla. Vaikeusasteen 2-3 vuotojen hoitoon tarvitaan usein immuunivastetta muuntavia lääkkeitä, useimmiten kortikosteroidia tai immunoglobuliini-infuusio. Prednisolonia annetaan joko 4 mg/kg/vrk 3-4 päivän ajan tai 1-2 mg/kg/vrk kahden viikon ajan ja immunoglobuliinia 0,8-1,0 g/kg kerta-annoksena (1). Immunoglobuliiniin saadaan nopeampi vaste, mutta se on kalliimpi ja hoito on vaikeampi antaa. Vaikeissa vuodoissa tarvitaan joskus myös punasoluinfuusioita. Trombosyyttisiirtoja annetaan vain harkiten, koska siirretyt trombosyytit tuhoutuvat ja vaikutus on lyhytkestoinen. Kontrolloimattomassa 4-5 asteen vuodossa tarvitaan eri hoitojen yhdistämistä ja mahdollisesti pernan poisto. Nykyään sekä Yhdysvalloissa että Euroopassa on hyväksytty suun kautta annettava trombopoietiinireseptorin agonisti eltrombopagi hoidoksi yli 1-vuotiaille lapsille, joilla on persistoiva ITP ja muilla hoidoilla saavutettu vaste on riittämätön tai olematon.
Lapsen liikkumista ja koulunkäyntiä rajoitetaan mahdollisimman vähän. Hänen tulee kuitenkin välttää harrastuksia, joissa voi saada suurienergiaisia vammoja, kuten ratsastusta ja laskettelua. Lisäksi vältetään trombosyyttifunktiota heikentäviä lääkkeitä, kuten tulehduskipulääkkeitä.
Ennenaikaisilla vauvoilla heti syntymän jälkeen havaittu trombosytopenia johtuu useimmiten antenataalisista syistä, kuten asfyksiasta, istukan vajaatoiminnasta tai kongenitaalisesta infektiosta (esim. CMV, toksoplasma tai coxsackievirus), ja se korjaantuu yleensä itsestään (15). Pienipainoisena syntyneillä ilmenee trombosytopeniaa, joka noin 90 %:lla osoittautuu ohimeneväksi ja etiologia jää epäselväksi (16). Neonataalinen alloimmuunitrombosytopenia (NAIT) aiheuttaa vastasyntyneelle vakavien vuotojen riskin, joten se on otettava huomioon jo diagnostiikan alkuvaiheessa varsinkin täysiaikaisilla ja vuoto-oireisilla vauvoilla. Trombosytopenia voi olla myös vakavan bakteeri-infektion tai keskosilla nekrotisoivan enterokoliitin aiheuttama. Jos vastasyntyneen trombosytopeniaan ei löydy syytä ja se pitkittyy yli kuukauden kestäväksi, on aktiivisesti etsittävä perinnöllisiä trombosytopeniaoireyhtymiä.
NAIT syntyy, kun äidissä kehittyy raskauden aikana vasta-aineita sikiön isältä perimiä trombosyyttispesifisiä antigeeneja kohtaan (human platelet antigen, HPA). Vasta-aineet läpäisevät istukan ja aiheuttavat sikiölle trombosytopenian, joka jatkuu, kunnes vasta-aineet ovat poistuneet elimistöstä. Yleisimmät trombosyyttiantigeenit ovat HPA-1, -2, -3, -4, -5, -6 ja -15, joista jokaisella on a- ja/tai b-alleeli joko homo- tai heterotsygoottisena. Tavallisin on anti-HPA-1a (75-80 %) ja toiseksi yleisin anti-HPA-5b (10-15 %) (17,18). Väestöstä 1,6-4,6 % on negatiivisia HPA-1a:n suhteen, mutta vain 10 %:lle heistä kehittyy anti-HPA-1a-vasta-aineita. Yleensä anti-HPA-1a-immunisaatio aiheuttaa vaikeamman taudin kuin anti-HPA-5b-immunisaatio.
Useimmiten NAIT on taudinkuvaltaan lievä, mutta toisaalta se on yleisin täysiaikaisen vastasyntyneen aivoverenvuodon aiheuttaja. Kallonsisäisen verenvuodon saa 7-26 % NAIT-vauvoista (19), ja 80 % aivoverenvuodoista tapahtuu jo kohdussa, noin puolet jo ennen raskausviikkoa 30. Aivoverenvuoto voi johtaa vammautumiseen ja kuolleisuus on 1-10 % (20). Toisin kuin punasoluista johtuva alloimmunisaatio, NAIT voi ilmetä jo ensimmäisessä raskaudessa. Uusimisriski seuraavissa raskauksissa on suuri ja yleensä taudinkuva vaikeampi. NAIT:n esiintyvyys vaihtelee kirjallisuudessa välillä 1/350-5 000 elävänä syntynyttä.
Äidin ITP:ssa trombosyyttivasta-aineita päätyy istukan kautta vauvaan, ja ne aiheuttavat myös vastasyntyneelle trombosytopenian (15). Myös äidin systeeminen lupus erythematosus voi aiheuttaa vastasyntyneen trombosytopeniaa. Niinpä jos vauvalla epäillään olevan NAIT, äidin ITP ja muut autoimmuunisairaudet on huolellisesti suljettava pois. NAIT varmistetaan tutkimalla äidin anti-HPA-vasta-aineet. Vanhempien ja vastasyntyneen HPA-antigeenit genotyypitetään. Vuoto-oireiselle vastasyntyneelle annetaan trombosyyttejä riippumatta trombosyyttitasosta ja tarvittaessa jo ennen kuin tyypitettyjä trombosyyttejä on tarjolla. Myöhemmin on parasta antaa HPA-tyypitettyjä trombosyyttejä. Ehkäisevä trombosyyttisiirto on tarpeellinen, jos trombosyyttitaso on alle 30 × 109/l. Pään kaikukuvaus on syytä toistaa varhain, mikäli trombosyyttitaso pysyy alle 50 × 109/l. Myös immunoglobuliini-infuusioon saadaan vastetta, mutta usein hitaammin kuin ITP:a hoidettaessa (15). Hätätilanteessa immunoglobuliini-infuusion voi antaa ennen tyypittämättömiä trombosyyttejä. Yleensä trombosyyttitaso normalisoituu 8-10 päivän ikään mennessä (21).
Kun NAIT-riski on tiedossa seuraavissa raskauksissa, äidille voidaan antaa ehkäisevästi immunoglobuliinia, steroidia tai intrauteriinisia trombosyyttisiirtoja (22). Intrauteriiniseen trombosyyttisiirtoon liittyy vakavan komplikaation riski ja invasiivisista toimenpiteistä pidättäytyminen on suositeltavaa (23).
Lääkkeet voivat aiheuttaa trombosytopenian heikentämällä verihiutaleiden tuotantoa tai lisäämällä niiden kulutusta. Solunsalpaajat aiheuttavat yleensä luuytimen ohimenevän lamaantumisen, joka tyypillisesti johtaa leuko- ja trombosytopeniaan. Useat lääkkeet saattavat aiheuttaa harvinaisena haittavaikutuksena luuytimen hypo- tai aplasian. Yleensä lääkkeen aiheuttamalla trombosytopenialla tarkoitetaan kuitenkin lääkkeen laukaisemaa immunologista trombosytopeniaa, jossa lääkeaineen tarttuminen verihiutaleen pintaan käynnistää vasta-ainetuotannon tai lisää jo olemassa olevan autovasta-aineen affiniteettia verihiutaleisiin (24).
Trombosytopenia alkaa tyypillisesti viikon kuluessa lääkkeen käytön aloittamisesta ja loppuu, kun lääke on poistunut elimistöstä. Miltei mikä lääke tahansa voi aiheuttaa trombosytopeniaa, mutta tyypillisimmin aiheuttajana on hepariini tai antibiootti (24,25). Lapsilla on kuvattu myös tulehduskipulääkkeiden ja parasetamolin aiheuttamia trombosytopenioita (26).
Myelodysplastinen oireyhtymä (MDS) on hematopoieettisten kantasolujen klonaalinen häiriö, joka johtaa luuytimen toimintahäiriöön ja aiheuttaa merkittävän riskin leukemian kehittymiseen (27,28). Lapsilla se ilmenee yleensä trombosytopeniana, neutropeniana ja anemiana. Se voi myös johtua perinnöllisestä luuytimen toimintahäiriöistä (esim. Fanconin anemia, dyskeratosis congenita ja GATA2-haploinsuffisienssi) tai kehittyä solunsalpaaja- tai sädehoidon jälkivaikutuksena (29). Etenevä myelodysplastinen oireyhtymä on allogeenisen kantasolusiirron aihe.
Vaikea aplastinen anemia (SAA) on harvinainen autoimmuuniprosessi, jossa kohteena ovat varhaisvaiheen multipotentit hematopoieettiset solut. Tämä johtaa vaikeaan pansytopeniaan (trombosytopenia, leukopenia ja anemia) ja luuytimen aplasiaan.
Hoito aloitetaan immunosuppressiivisella lääkityksellä (30). Yleensä potilaat tarvitsevat toistuvia trombosyytti- ja punasolusiirtoja. Mikäli luuydin ei toivu, tehdään allogeeninen luuytimen kantasolusiirto.
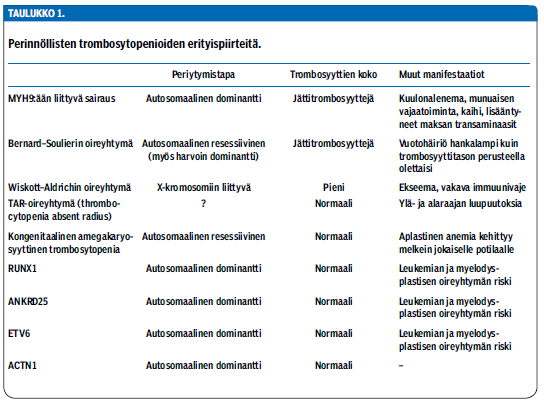
Synnynnäisellä trombosytopenialla tarkoitetaan yleensä perinnöllistä vikaa trombosyyttien tuotannossa. Geenitutkimus on mullistanut perinnöllisten trombosytopenioiden diagnostiikan: aikaisemmin kuvatut sairaudet on määritelty ja ryhmitelty geneettisin perustein ja kokonaan uusia trombosytopeniasairauksia on kuvattu.
Perinnöllinen trombosytopenia on harvinainen, ja sen aiheuttajamutaatioiden joukko on varsin laaja. Joskus trombosyyttien koko, dysmorfiset piirteet, muut elinmanifestaatiot ja periytymistapa johtavat oikean diagnoosin jäljille (taulukko 1 «»1), mutta usein eteenpäin päästään vain geenipaneelitutkimuksilla. Nykyisin tunnetaan ainakin 22 perinnöllisen trombosytopenian geenitausta (13), mutta edelleen noin puolet ilmeistä perinnöllistä trombosytopeniaa sairastavista jää ilman geneettistä diagnoosia. Toisaalta vaikuttaa siltä, että hankalimmat hoitoa tai seurantaa vaativat geenimutaatiot sisältyvät valtaosin jo nyt käytössä oleviin geenipaneeleihin.
Yleisin perinnöllinen trombosytopenia on MYH9:ään liittyvä sairaus (MYH9-RD) (31,32). Se periytyy autosomissa vallitsevasti ja johtuu MYH9-geenin (myosin heavy chain 9) mutaatiosta.
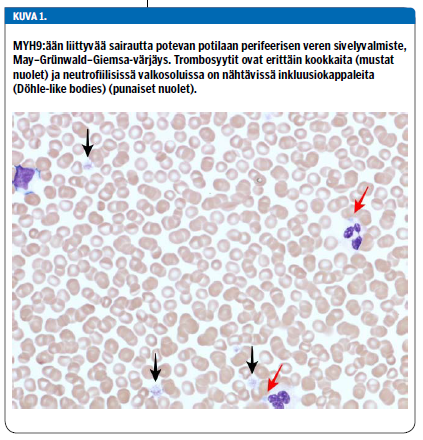
Sairauden tyypillinen piirre ovat valtavan kookkaat, harvalukuiset trombosyytit (kuva 1 «»3). Trombosyyttiluku on syntymästä asti ja vakaasti poikkeavan pieni, mutta vaihtelee jopa samaa mutaatiota kantavilla potilailla välillä 10-150 × 109/l. Vuototaipumus on yleensä lievä, mutta kaikkein matalimpiin trombosyyttitasoihin liittyy merkittävien vuotojen riski.
Kaikilla potilailla on trombosytopenia, mutta sen lisäksi tautiin liittyy muita manifestaatioita. Noin 60 %:lle kehittyy yleensä kuulolaitetta edellyttävä, molemminpuolinen sensorineuraalinen kuulonalenema ja noin 30 %:lle munuaisten vajaatoiminta, joka vaatii usein dialyysihoitoa. Lisäksi 10-16 % potilaista saa kaihin. Potilailla esiintyy myös poikkeavan suuria maksan transaminaasiarvoja, mutta oireyhtymään liittyviä maksan toimintahäiriöitä ei ole kuvattu.
MYH9:ään liittyvä sairaus diagnosoidaan osoittamalla immunovärjäyksellä veren neutrofiileistä myosiiniaggregaatteja (non-muscle myosin heavy chain II) tai tutkimalla MYH9-geenin mutaatio. Kun mutaatio tunnetaan, eri elinmanifestaatioiden riski voidaan arvioida varsin luotettavasti (33).
Bernard-Soulierin oireyhtymän (BSS) aiheuttaa vika von Willebrandin tekijän reseptorissa (34), joko reseptorin puuttuminen solun pinnalta tai reseptorin viallinen toiminta. Tauti periytyy yleensä autosomissa resessiivisesti. Trombosyytit ovat normaalia selvästi kookkaampia. Vaikka trombosytopenia on suhteellisen lievä (keskimääräinen trombosyyttimäärä on 60 × 109/l), von Willebrandin tekijän reseptorivian vuoksi potilailla on keskivaikea vuototaipumus.
Oireyhtymä on kansainvälisesti harvinainen sairaus (n. 1 potilas miljoonaa synnytystä kohden), mutta Suomessa se on jonkin verran yleisempi. Resessiivisessä muodossa trombosyyttien aggregaatiotutkimuksessa ristosetiinivaste puuttuu. Diagnoosi varmistetaan virtaussytometrisesti tai geenitestillä.
Wiskott-Aldrichin oireyhtymän (WAS) ja sen lievemmän muodon, X-kromosomiin liittyvän trombosytopenian (X-linked thrombocytopenia, XLT) aiheuttaa X-kromosomissa sijaitsevan WAS-geenin mutaatio (35). Potilailla on miltei aina trombosytopenia, mutta sen vaikeusaste vaihtelee runsaasti ja vuototaipumus merkittävästä hyvin lievään. Trombosyytit ovat pienikokoisia.
Wiskott-Aldrichin oireyhtymä aiheuttaa myös ekseemaa ja vakavan immuunivajeen. Immuunivajeen vuoksi se on aihe luuytimen kantasolusiirrolle (36). X-kromosomiin liittyvä trombosytopenia on huomattavasti lievempi sairaus, jota voidaan yleensä turvallisesti vain seurata.
Thrombocytopenia absent radius eli TAR on harvinainen oireyhtymä, jossa trombosyyttejä on synnynnäisesti niukasti (yleensä alle 50 × 109/l) ja potilailla on lisäksi yläraajoihin painottuvia luupuutoksia, jotka vaihtelevat radiuksen puuttumisesta jopa koko yläraajan puuttumiseen (37). TAR:n periytyminen on monimutkaista ja tunnetaan edelleenkin huonosti, mutta suuri osa tapauksista liittyy kromosomialueen 1q21.1 mikrodeleetioihin.
Kongenitaalinen amegakaryosyyttinen trombosytopenia (CAMT) on harvinainen etenevä luuytimen toimintahäiriö, joka johtuu trombopoietiinin reseptorigeenin mutaatiosta (38). Potilailla on jo vastasyntyneenä syvä trombosytopenia, joka etenee aplastiseksi anemiaksi yleensä ensimmäisten elinvuosien aikana. Tautiin ei liity dysmorfioita tai muita oireyhtymäpiirteitä. Luuytimen kantasolusiirto on parantava hoito.
Viime vuosina on kuvattu kolme autosomissa vallitsevasti periytyvää mutaatiota, jotka kaikki aiheuttavat merkittävän trombosytopenian ja selvästi lisääntyneen leukemiariskin. Näille mutaatioille on myös tyypillistä, että trombosyytit ovat normaalikokoisia.
RUNX1-geenin mutaatio aiheuttaa yli 90 %:lle kantajista trombosytopenian ja joka toiselle ekseeman. Mutaatioon liittyvä myelodysplastisen oireyhtymän tai akuutin myelooisen leukemian riski on noin 40 % (39,40). Mutaatio ANKRD25-geenissä aiheuttaa trombosytopenian ja arvioilta 10-20-kertaisen leukemia- ja MDS-riskin (41). ETV6-geenin mutaatio aiheuttaa trombosytopenian lisäksi punasolujen makrosytoosia, ja joka neljäs mutaation kantaja sairastuu leukemiaan (42).
Mutaatio aktiinia koodaavassa ACTN1-geenissä aiheuttaa autosomissa vallitsevasti periytyvän trombosytopenian, jossa trombosyytit ovat poikkeuksellisen kookkaita (43) Tähän mutaatioon liittyy lievä trombosytopenia ja vuototaipumus ilman muita elinmanifestaatiota.
Perinnöllisissä trombosytopenioissa vuoto-ongelmat ovat yleensä varsin vähäisiä. Potilaiden on syytä välttää verihiutaleiden toimintaa heikentäviä lääkkeitä, kuten tulehduskipulääkkeitä. Oireenmukaiseksi hoidoksi voidaan käyttää traneksaamihappoa. Runsaita kuukautisia voidaan hoitaa yhdistelmäehkäisypillereillä, mutta ennen hoidon aloittamista on käytävä huolellisesti läpi tromboosin riskitekijät, sillä perinnöllisiin trombosytopenioihin ja erityisesti kookkaisiin verihiutaleisiin voi liittyä myös tromboosialttiutta.
Synnynnäiseen trombosytopeniaan liittyvä vuotoalttius voidaan korjata nopeasti trombosyyttisiirrolla. Näin on syytä toimia esimerkiksi ennen kirurgisia toimenpiteitä. Ennen suuria leikkauksia olisi pyrittävä trombosyyttitasoon yli 50 × 109/l ja kriittisille vuodoille altistavissa leikkauksissa, kuten neuro- ja silmäkirurgiassa, tasoon yli 100 × 109/l (13).
Usein trombosytopeniaoireyhtymissä muut kuin hematologiset ongelmat ovat merkittävämpiä kuin trombosytopenia. Siksi seurannan järjestäminen yhteistyössä lääketieteen eri erikoisalojen kanssa on tärkeää. Perinnöllistä trombosytopeniaa sairastava on myös ohjattava perinnöllisyysneuvontaan.
Käytännössä lääkärin suurin haaste on erottaa yleinen ja hyvänlaatuinen ITP ja harvinaisemmat trombosytopenian syyt, jotka edellyttävät seurantaa tai välittömiä toimenpiteitä. Lapsuusiän ITP-potilas on yleensä alle kouluikäinen ja hyväkuntoinen, vuoto-oireet ovat vähäisiä ja verenkuva on trombosytopeniaa lukuun ottamatta normaali. Usein potilas on myös sairastanut hiljattain infektion tai saanut rokotteen. Huonokuntoinen lapsi, jolla on trombosytopenia, on syytä lähettää päivystyspotilaana erikoissairaanhoidon arvioon, kuten myös potilaat, joilla on useamman verisolulinjan poikkeavuus. Myös lapsipotilaat, joilla on vuoto-oireinen ITP, kuuluvat erikoislääkärin hoitoon.
Jos potilaalta aikaisemmin mitattu trombosyyttiarvo on ollut normaali, suvussa ei esiinny poikkeavan matalia trombosyyttiarvoja eikä potilaalla todeta dysmorfisia piirteitä tai muita elinmanifestaatiota, perinnöllinen trombosytopenia on epätodennäköinen. Mikäli epäily synnynnäisestä trombosytopeniasta herää, on sairauden etiologia syytä selvittää, vaikka potilas olisi oireeton.
A child with thrombocytopenia is a common problem for the general practitioner. Typically, a low platelet count is due to immune thrombocytopenia (ITP), which has an excellent prognosis among children and a low risk of serious bleeding. However, a low platelet count can also be a sign of severe infection or leukaemia. A child who is in a poor general condition or who has alterations in multiple blood cell lineages should be immediately referred to hospital. Children with persistent or chronic ITP and patients with significant bleeding symptoms should also be referred to a paediatrician or a paediatric haematologist. Congenital thrombocytopenias are rare conditions, but in the case of persistently low platelet counts with other syndromic symptoms or suspected inherited thrombocytopenia, genetic testing should be considered. Diagnosis of inherited thrombocytopenia is important in order to spare the patient from futile attempts to treat ITP and to arrange adequate follow-up.