

Ks. artikkelin pdf-versio «http://www.fimnet.fi/cl/laakarilehti/pdf/2019/SLL422019-2353.pdf»1 Lääkärilehden sivuilla (vaatii FiMnet-tunnuksen).
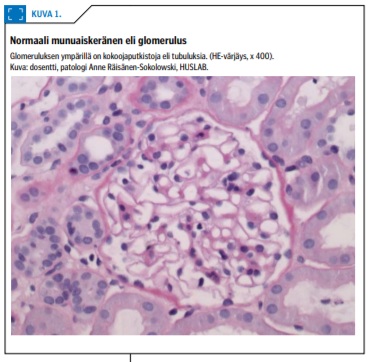
Glomerulus eli munuaiskeränen on munuaisten suodatusyksikön keskeinen osa: siinä primaarivirtsa suodattuu kapillaariverisuonista ja jatkaa kokoojaputkistoon eli tubulukseen (kuva 1 «»1). Munuaistautia, joka vaurioittaa pääasiassa glomeruluksia, kutsutaan glomerulonefriitiksi eli munuaiskerästulehdukseksi.
Diagnostiikassa keskeinen rooli on munuaiskoepalan valomikroskooppisella löydöksellä yhdistettynä immunofluoresenssitutkimukseen. Joskus tarvitaan myös elektronimikroskopiaa (1). Glomerulonefriitin nimi juontuu yleisimmin suoraan patologis-anatomisesta löydöksestä.
Osa glomerulonefriiteistä saattaa vaurioittaa vain joitakin glomeruluksia (fokaalinen muutos), osa kaikkia (diffuusi muutos). Globaalinen vaurio käsittää koko glomeruluksen ja segmentaalinen puolestaan vain osan yksittäisestä keräsestä.
Suomessa glomerulonefriittien ilmaantuvuus on noin 18/100 000, ja dialyysiin potilaista etenee alle 10 % (2). Munuaistautirekisterin tuoreimman raportin perusteella glomerulonefriitti on kuitenkin meillä neljänneksi yleisin munuaisten loppuvaiheen vajaatoiminnan syy (3).
Glomerulonefriittien syntymekanismi vaihtelee, mutta taustalla on useimmiten immunologisella mekanismilla ilmaantuva kudosvaurio, jossa geneettisillä ja ympäristötekijöillä on vaihteleva merkitys (4). Primaariset taudit rajoittuvat munuaisiin, mutta glomerulusvaurio voi olla myös osa systeemisen sairauden ilmentymää, esimerkiksi vaskuliittien yhteydessä. Tässä katsauksessa käydään läpi tärkeimmät primaariset glomerulonefriitit.
Glomerulonefriitin diagnoosi edellyttää munuaiskoepalan tutkimista, eikä eri glomerulonefriittejä voi luotettavasti erotella toisistaan kliinisin perustein (5). Pelkkä mikroskooppinen hematuria ei ole enää vuosiin ollut munuaisbiopsian aihe, ellei epäillä jotakin systeemistä tautia, jossa histologisen diagnoosin saaminen on tärkeää. Mikäli koepalan ottoon ei ryhdytä, potilasta tulee seurata määrävälein. Jatkossa koepala otetaan, jos tutkimisen aiheet täyttyvät seurannassa eikä vasta-aiheita ole (taulukko 1 «»2).

Kliininen ilmiasu voi antaa vihjeitä munuaistaudin taustasta. Verenpaine on suurimmalla osalla potilaista koholla jo diagnosointivaiheessa. Potilaalla voi olla selviä oireita (taulukko 2 «»3) tai hän voi olla täysin oireeton ja selvittely käynnistyy sattumalta todetusta poikkeavuudesta (5).

Glomerulonefriiteissä on yleisimmin aktiivinen virtsalöydös eli hematuriaa ja sen lisäksi vaihtelevan asteista proteinuriaa. Nefriittisestä sedimentistä puhutaan, kun on hematuria ja virtsassa usein myös punasolulieriöitä. Pääosa virtsaan erittyvästä proteiinista on albumiinia ja osalle potilaista voi kehittyä nefroottinen oireyhtymä eli turvotuksia ja hypoalbuminemia yhdistyneenä runsaaseen proteinuriaan ((≥ 3-3,5 g/vrk). Munuaisten toiminta voi olla joko normaali tai vaikeastikin häiriintynyt. Taulukossa 3 «»4 on lueteltu perusdiagnostiikassa tarvittavat tutkimukset.

Hitaasti lisääntyvä proteinuria, hematuria ja heikentyvä munuaisten toiminta sopivat moniin glomerulonefriitteihin (esim. IgA-glomerulonefriitti), kun taas akuutti, nopeasti etenevä munuaisten vajaatoiminta aktiiviseen virtsalöydökseen yhdistyneenä sopii esimerkiksi vaskuliittiin (5).
Hoito valitaan glomerulonefriitin aiheuttajan ja kliinisen taudinkuvan perusteella. Yhteisiä hoitoja eri taudeissa ovat verenpaineen, proteinurian ja turvotusten hoito sekä munuaisten vajaatoiminnan kehityttyä sen seurausten hoito (1). Eräillä verenpainetta alentavilla lääkkeillä on myös itsenäinen proteiinin erittymistä virtsaan hillitsevä vaikutus. Tavallisimmat glomerulonefriittien hoidossa tarvittavat lääkitykset ja hoidon kohteet on esitetty taulukossa 4 «»5.
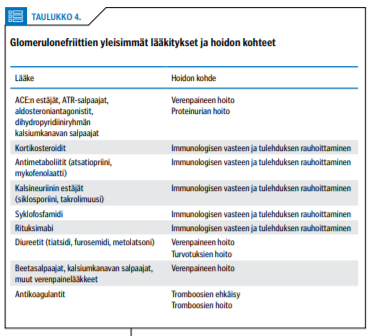
Tulehduksellista komponenttia hillitään immunomoduloivalla lääkityksellä, ja sen aloittamisen aiheet vaihtelevat taudeittain (5). Nopeasti etenevissä glomerulonefriiteissä riittävän varhainen ja tehokas immunosuppressiivinen hoito parantaa munuaistaudin ennustetta ratkaisevasti (6). Hitaasti etenevissä tautimuodoissa ei välttämättä tarvita immunomoduloivaa hoitoa lainkaan. Nefroottiseen oireyhtymään liittyy erityisesti membranoottisessa glomerulonefriitissä suurentunut laskimotukosriski (5,7).
Verenpainetauti, heikentynyt munuaisten toiminta ja runsaana jatkuva proteinuria ovat useimmissa glomerulonefriiteissa vajaatoiminnan etenemisen itsenäisiä riskitekijöitä (5). Samoin eräät histologiset löydökset, kuten runsaat krooniset vauriot glomeruluksissa tai välikudoksessa, ennakoivat vajaatoiminnan etenemistä.
IgA-glomerulonefriitti (IgAGN) on maailman yleisin primaarinen glomerulonefriitti. Sen vuotuinen ilmaantuvuus Suomessa on noin 5/100 000 (2). Tautia esiintyy miehillä enemmän kuin naisilla, ja esiintyvyydessä on lisäksi selviä eroja maailmanlaajuisesti. Tavallisin toteamisikä on 20-40 vuotta, mutta tauti voi ilmaantua missä iässä tahansa.
Taudin perimmäinen syy on edelleen osittain selvittämättä. Sairastuneilla on todettu poikkeavuuksia IgA-immunoglobuliinissa. Sopivan lisätekijän ilmaantuessa (mahdollisesti jokin mikrobi, ravinnon antigeeni tai tunnistamaton ympäristötekijä) käynnistyy siihen kohdistuva vasta-ainetuotanto. Sen seurauksena syntyvät immunokompleksit sakkautuvat glomeruluksiin aiheuttaen tulehduksen ja lopulta kliinisen taudin. Suurin osa tapauksista on sporadisia, mutta potilaiden terveillä lähisukulaisilla on todettavissa samoja IgA-immunoglobuliinin poikkeavuuksia kuin potilailla (8).
Diagnoosi perustuu munuaiskoepalan immunofluoresenssitutkimukseen, jossa todetaan tyypillinen IgA-kertymä. Valomikroskooppinen löydös voi olla vaihteleva.
Tyypillisin taudinkuva on oireettoman potilaan sattumalta todettu proteinuria ja hematuria yhdistyneenä kohonneeseen verenpaineeseen. Hieman alle puolella potilaista virtsa voi kuumetautien alussa ohimenevästi muuttua makroskooppisesti veriseksi. Pienellä osalla potilaista proteiinnin erittyminen virtsaan voi alkaa hyvin äkillisesti ja olla erittäin runsasta, samaan tapaan kuin vähämuutoksisessa eli minimal change -glomerulonefriitissä, tai munuaisten vajaatoiminta voi edetä hyvin nopeasti ja taudinkuva muistuttaa vaskuliittia (8).
IgA-glomerulonefriitti voi olla myös osa systeemistä tautia, jossa ilmaantuu ihon purppuramuutoksia ja vaihtelevasti niveloireita ja vatsakipuja. Tällöin puhutaan IgA-vaskuliitista (aiemmin Henoch-Schönleinin purppura) (9). Se on pienten suonten vaskuliitti, jossa todetaan IgA-saostumia oireilevien elinten hiussuonissa ja munuaismanifestaatio noin puolella potilaista. Munuaiskoepalan löydös on identtinen primaarisen IgA-glomerulonefriitin löydöksen kanssa, ja taudin nimi ratkeaakin yleisoireiden perusteella. IgA-vaskuliitti on selvästi yleisempi lapsilla, mutta sitä tavataan myös aikuisväestössä. Siihen liittyvän munuaistaudin hoito noudattelee IgA-glomerulonefriitin hoitolinjoja (10).
Keliakiaa, maksasairauksia, tulehduksellista suolistosairautta ja sidekudostauteja sairastavilla on raportoitu esiintyvän IgA-glomerulonefriittiä enemmän kuin pelkän sattuman aiheuttamana olettaisi. Syy-seuraussuhteet eivät läheskään aina ole täysin selviä. Hoito näissä yhteyksissä ei näytä olennaisesti poikkeavan primaaristen tautimuotojen hoidosta (11). Kiinnostavia yksittäisraportteja on kuitenkin julkaistu mm. keliakiapotilaan gluteenittoman ruokavalion aikaansaamasta munuaistaudin kliinisestä remissiosta (12).
Parantavaa hoitoa IgA-glomerulonefriittiin ei ole, mutta pienellä osalla potilaista taudin kliiniset löydökset saattavat hävitä ilman hoitotoimia. Munuaisten loppuvaiheen vajaatoiminta kehittyy 20-25 vuoden seurannassa 25-30 %:lle potilaista. Hoidon kulmakiviä ovat proteinuriaa vähentävä lääkitys sekä tehokas verenpaineen hoito erityisesti ACE:n estäjillä, ATR-salpaajilla tai molemmilla. Tavoitteena on saada proteinuria alle 1 g:aan/vrk ja verenpaine jopa alle 125/75 mmHg:iin. Lääkitys on tarpeen myös normotensiivisille, jos proteinuriaa esiintyy. Tuplasalpausta ACE:n estäjän ja ATR-salpaajan yhdistelmällä voidaan harkita valikoiduille potilaille, mutta siihen voi liittyä hyperkalemian ja munuaisten vajaatoiminnan pahentumisen riski.
Puolen vuoden glukokortikoidihoitoa harkitaan, mikäli proteinuria pysyy yli tavoitteiden optimoidusta muusta hoidosta huolimatta ja eGFR on vielä riittävän hyvä (tavallisesti > 30 ml/min) (1,8,10). Hoitopäätöksessä voidaan nojata histologisiin löydöksiin, erityisesti rajatapauksissa. Glukokortikoidihoidon toistamisen hyödyllisyydestä hitaasti etenevässä tautimuodossa ei ole näyttöä.
Tauti voi uusiutua myös munuaissiirrännäiseen.
Vähämuutoksinen eli minimal change -glomerulonefriitti (MCNS) löytyy taustalta jopa 90 %:ssa 1-10-vuotiaiden lasten ja 10-15 %:ssa aikuisten nefroottisista oireyhtymistä. Lapsilla vuosittainen ilmaantuvuus on 2-7 uutta tapausta 100 000 henkilöä kohti, ja tauti on pojilla yleisempi (2 : 1) (13), Suomessa ilmaantuvuus on 1-2/100 000 (2).
Glomerulusten tyvikalvon jalkalisäkkeet (podosyytit) vaurioituvat. Laukaisevaksi tekijäksi on ehdotettu lukuisia molekyylejä (mm. interleukiini 13). T-solujen ja B-solujen toimintahäiriöillä lienee myös merkitystä taudin synnyssä (14,15).
Suurin osa tapauksista on idiopaattisia. Sekundaarisen taudin voivat aiheuttaa monet lääkkeet (tulehduskipulääkkeet, antibiootit, litium), syövät (erityisesti hematologiset), infektiot (tuberkuloosi, mykoplasma, C-hepatiitti), autoimmuunitaudit ja allergiat. Taudinkuvassa tyypillistä on äkillisesti muutamien päivien kuluessa alkava runsas proteinuria, jonka oireena ovat kudosturvotukset. Hankalimmillaan nestettä kertyy sekä vatsaonteloon että sydän- ja keuhkopussiin (15). Mikroskooppista hematuriaa tavataan 20 %:lla, lisäksi hypoalbuminemia ja hyperlipidemia ovat tyypillisiä löydöksiä. Osalla potilaista myös plasman kreatiniinitaso on koholla.
Munuaisten vajaatoimintaa tavataan diagnosointivaiheessa 20-30 %:lla aikuispotilaista. Sen riskiä lisäävät kohonnut verenpaine, miessukupuoli, vaikea nefroosi ja korkea ikä. Yleensä taustalla on korjaantuva munuaisvaurio (14,15).
Taudin yleisyyden vuoksi munuaiskoepala tutkitaan lapsilta vasta, jos tyypillisen taudinkuvan yhteydessä ei saada vastetta kortisonihoitoon. Aikuisilla diagnoosi perustuu koepalaan ennen hoitojen aloittamista. Valomikroskooppinen ja immunofluoresenssitutkimus ovat yleensä normaalit (josta taudin nimi minimal change). Elektronimikroskopiassa nähdään jalkalisäkkeiden yhteensulautuminen.
Hoito aloitetaan yleensä suuriannoksisella kortisonilla suun kautta. Steroidille herkistä lapsista jopa 80-90 % saa jossain vaiheessa ainakin yhden uusiutuman, aikuisista 50-75 %. Useasti uusiutuva tai steroidiriippuvainen tautimuoto voi vaatia toistuvia kortisonikuureja ja steroidin korvaamista muilla tulehdusta rauhoittavilla valmisteilla (taulukko 4 «»5). Steroidille resistentiksi tauti luokitellaan, jos lapselle ei saada vastetta 4-8 viikossa ja aikuiselle 16 viikossa. Osalle näistä potilaista saadaan hoitovaste muilla tulehdusta rauhoittavilla valmisteilla (14,15).
Jos proteinuria saadaan hallintaan, taudin ennuste on hyvä eikä pysyvää munuaisten vajaatoimintaa juuri kehity. Huonoin ennuste on steroidille resistentissä taudissa. Näiden potilaiden uusintakoepaloissa todetaan usein muu glomerulonefriitti (fokaalinen segmentaalinen glomeruloskleroosi).
Fokaalisen segmentaalisen glomeruloskleroosin (FSGS) vuosittainen ilmaantuvuus on 0,2-1,8/100 000, ja tauti on miehillä 1,5 kertaa tavallisempi kuin naisilla (16).
Taudille on tyypillistä samankaltainen jalkalisäkevaurio kuin minimal change -glomerulonefriitissä, ja onkin esitetty, että kyseessä ovat saman taudin eri muodot (17). Jalkalisäkkeiden tuhoutumista seuraa parietaalisten epiteelisolujen aktivoituminen, joka johtaa lopulta kapillaarien tukkeutumiseen. Tutkimuksissa on pyritty tunnistamaan jalkalisäkevaurion ja parietaalisolujen aktivaation merkkiaineita (mm. liukoinen urokinaasireseptori suPAR), jotta minimal change -glomerulonefriitti ja fokaalinen segmentaalinen glomeruloskleroosi voitaisiin erottaa toisistaan. Toistaiseksi mikään tutkituista merkkiaineista ei ole päätynyt kliiniseen käyttöön (16,18).
Biopsiassa tyypillistä on fokaalinen ja segmentaalinen mesangiaalisen matriksin lisääntyminen, kapillaarien häviäminen, skleroosi, hyalinoosi ja arpeutuminen. Immunofluoresenssitutkimuksessa IgM ja C3 ovat positiivisia. Elektronimikroskopia varmistaa jalkalisäkkeiden yhteensulautumisen, primaarisessa taudissa laajasti, sekundaarisessa vähemmän (19,20).
Primaarisen fokaalisen segmentaalisen glomeruloskleroosin aiheuttajaa ei tunneta. Teoriaa kiertävän, jalkalisäkkeisiin vaikuttavan tekijän olemassaolosta tukee alttius taudin nopeaan uusiutumiseen munuaissiirteessä. Tauti alkaa nopeasti ja potilaat ovat yleensä nefroottisia, hematuriaa tavataan noin puolella, ja kreatiniinitaso on koholla 25-50 %:lla. Hoito perustuu immunosuppressioon (taulukko 4) (16,21). Primaarinen tauti uusii munuaissiirteessä 30-50 %:lla potilaista. Remissio voidaan saada aikaan plasmanvaihdoilla, suuriannoksisella siklosporiinilla ja rituksimabilla (22).
Fokaalisesta segmentaalisesta glomeruloskleroosista tunnetaan lukuisia geneettisiä muotoja. Tärkeimmät näistä ovat nefriinin (NPHS1), podosiinin (NPHS2) ja fosfolipaasi C epsilon 1:n (PLCe1) mutaatiot. Geenitestausta suositellaan, kun potilas on alle 1-vuotias tai kun isomman lapsen nefroottisessa oireyhtymässä ei saada vastetta kortisonihoitoon. Positiivinen sukuanamneesi tai laajemman oireyhtymän epäily puoltavat geeniselvityksiä nuorille aikuisillekin. Geneettinen tauti on yleensä steroidille resistentti, ja kalsineuriinin estäjälääkitys tehoaa harvoille (16,21). Hiljattain on kuvattu myös apolipoproteiini L1 -geeniin (APOL1) assosioituva tautimuoto, joka on yleinen etenkin tummaihoisilla (23).
Sekundaarinen fokaalinen segmentaalinen glomeruloskleroosi on usein vaste glomerulusten kuormitumiseen mm. ylipainon tai vähentyneen munuaiskudoksen seurauksena (esim. anomaliat, munuaisen poisto). Se voi olla myös lääkkeiden (interferoni, bisfosfonaatit), virusten (HIV, sytomegalovirus) ja anabolisten steroidien tai heroiinin käytön aiheuttama. Sekundaarinen tauti ei yleensä ole nefroottinen. Hoidon perusta on proteinuriaa hillitsevä ja verenpainetta alentava lääkitys. Näyttöä immunosuppressiivisen lääkityksen hyödyistä ei ole. Mahdollinen virustauti tulee hoitaa ja kuormittavien valmisteiden käyttö lopettaa (16,19).
Ennusteen kannalta proteinurian vähentäminen on ensisijaista. Fokaalinen segmentaalinen glomeruloskleroosi johtaa munuaisten loppuvaiheen vajaatoimintaan 40-70 %:lla 10-20 vuoden kuluessa diagnoosista (16,22).
Membranoottinen glomerulonefriitti (MN) on nefroottista oireyhtymää aiheuttavista glomerulonefriiteistä yleisimpiä aikuisväestössä. Sen vuotuinen ilmaantuvuus Suomessa on 1-2/100 000 (2). Se ilmenee keskimäärin 50-60 vuoden iässä ja on miehillä kaksi kertaa niin yleinen kuin naisilla. Lapsilla tauti on harvinainen (24).
Noin 20 %:lla potilaista taudin taustalta löytyy jokin laukaiseva tekijä. Tällaisia ovat mm. useat autoimmuunisairaudet (esim. systeeminen lupus erytematosus, SLE), krooniset infektiot (esim. virushepatiitit), lääkkeet (kulta, penisillamiini, tulehduskipulääkkeet), syövät (kiinteät kasvaimet ja hematologiset syövät). Lopuilta laukaisevaa tekijää ei löydy, ja heillä tauti on primaarinen.
Tautiprosessi käynnistyy, kun jalkalisäkkeiden alapinnalle kertyy toimintaa häiritsevää IgG-vasta-ainetta. Sen muodostamat immunokompleksit johtavat lopulta proteinurian kehittymiseen. Primaarista tautia sairastavista noin 70 %:lla on veressä vasta-aineita M-tyypin fosfolipaasi A2 -reseptoria (PLA2R) vastaan (25). Se on tyvikalvon proteiinirakenne, jonka fysiologista tehtävää ei tunneta. PLA2R-vasta-aineet ovat hyvin spesifisiä, eikä niitä juurikaan esiinny sekundaarisen tautimuodon tai muiden munuaistautien yhteydessä. Onkin esitetty, että osalla potilaista voitaisiin luopua munuaisbiopsiasta kokonaan, jos PLA2R-vasta-aineet ovat positiiviset eikä ole kliinistä epäilyä muusta sairaudesta (1). Tautiin liittyviä antigeeneja löytynee tulevaisuudessa lisää.
Tyypillinen löydös on nefroottinen oireyhtymä, ja hematuriaa esiintyy puolella potilaista. Verenpaine on koholla noin 30 %:lla. Alkuvaiheessa kreatiniinitaso on usein normaali tai lievästi koholla. Ei-nefroottisilla potilailla munuaistoiminnan säilymisen ennuste on hyvä, mutta osalla potilaista runsas nefroosi säilyy, ja munuaisten loppuvaiheen vajaatoiminta kehittyy 10 vuoden kuluessa noin 35 %:lle (24).
Biopsiassa nähdään glomerulustyvikalvoilla ns. "piikkejä" ja tyvikalvoilla IgG:tä, joka primaarisessa taudissa on enimmäkseen IgG4:ää. Samassa lokalisaatiossa nähdään positiivinen PLA2-reseptorivärjäytyvyys.
Hoidossa noudatetaan aiemmin mainittuja hoitoperiaatteita. Profylaktista antikoagulaatiohoitoa suositellaan, kun potilas on nefroottinen. Sekundaarisessa taudissa hoito kohdistuu laukaisevaan tekijään. Primaarista tautia sairastaville potilaille tulee harkita immunomoduloivaa hoitoa (taulukko 4 «»5), jos munuaisten vajaatoiminnan kehittymisen riski on suuri (24). PLA2R-vasta-ainepositiivisilla vasta-ainetaso toimii hyvänä seurantamittarina: hoidon onnistuessa vasta-aineet vähenevät tai jopa häviävät, ja tämä johtaa yleensä viiveellä proteinurian rauhoittumiseen. Uudelleen ilmaantuvat vasta-aineet voivat ennakoida kliinistä uusiutumaa (1,24).
Yleislääkärin tehtäväksi jää usein lähinnä munuaistautien tunnistaminen, ja siihen perustyökalut riittävät mainiosti. Munuaisten toimintaa kuvaavat mittarit, tavallisimmin veren kreatiniini yhdistettynä laskennalliseen glomerulussuodosnopeuteen (eGFR) ja/tai veren kystatiini C, ovat käyttökelpoisia, ja myös arvio virtsan poikkeavuuksista kuuluu perustason tutkimuksiin. Jos potilaalla on hematuriaa kemiallisen seulonnan perusteella, se on varmennettava solutarkastelulla. Mikäli virtsaseula paljastaa proteinurian, sen määrä on syytä tutkia. Munuaisten kaikukuvaus kuuluu myös epäselvän munuaislöydöksen perustutkimuksiin.
Kiitämme dosentti, osastonylilääkäri, patologi Anne Räisänen-Sokolowskia munuaisen histologisesta kuvasta.
Glomerulonephritis is the fourth commonest reason for end-stage renal disease in Finland. Assessment of kidney function and the amount of proteinuria and haematuria are crucial elements when glomerulonephritis is suspected. The cornerstone of the diagnosis is renal biopsy. The general principles in the management of glomerular disease include treatment of hypertension and proteinuria reduction. Some patients require immunosuppressive treatment. This review covers the basic elements of the most common primary glomerulonephritis.