

Ks. artikkelin pdf-versio «http://www.fimnet.fi/cl/laakarilehti/pdf/2022/SLL3-4-2022-134.pdf»1 Lääkärilehden sivuilla (vaatii FiMnet-tunnuksen).
Epilepsiaa sairastaa lähes 1 % väestöstä (1,2). Sen etiologia voi olla geneettinen, rakenteellinen, infektioperäinen, aineenvaihdunnallinen, immuunivälitteinen tai tuntematon (3). Geneettinen epilepsia aiheutuu tunnetusta tai oletetusta geneettisestä muutoksesta. Se ei ole välttämättä sama kuin perinnöllinen epilepsia, sillä epilepsian aiheuttavat geneettiset muutokset ovat usein uusia ns. de novo -mutaatioita.
Myöskään geneettisesti monitekijäiset epilepsiat eivät yleensä ole selkeästi perinnöllisiä. Epilepsian rakenteellinen etiologia voi olla myös geenivirheen aiheuttama, kuten tuberoosiskleroosissa tai osassa aivojen kuorikerroksen kehityshäiriöitä.
Epileptinen enkefalopatia tarkoittaa varhaislapsuudessa alkavaa tilaa, jossa epileptinen häiriö aiheuttaa kehityksen ja käyttäytymisen poikkeavuutta enemmän kuin taustalla olevan etiologian yksistään voisi olettaa aiheuttavan. Epilepsiaa selittäviä geenimuutoksia on löytynyt erityisesti vaikeiden kehityksellisten sairauksien taustalta (4,5). Niihin liitettyjä tautimekanismeja ovat esimerkiksi ionikanavien sekä synaptisten proteiinien ja reseptorien toimintahäiriöt, muutokset solujen kasvussa ja proliferaatiossa sekä geeniekspression säätelyssä.
Dravet'n oireyhtymä (6,7) on tällä hetkellä tutkituin epileptinen enkefalopatia. Sen ilmaantuvuus ensimmäisen ikävuoden aikana on 5:100 000. Sairauden syy löydetään nykymenetelmin yli 80 %:lta potilaista. Mutaatio on yleisimmin natriumionikanavaa koodittavassa SCN1A -geenissä. Kyseessä on lähes aina uusi mutaatio.
Tyypillinen ensioire on yli 15 minuuttia kestävä tajuttomuus-kouristuskohtaus kuumeen yhteydessä. Kohtausoireet ovat monimuotoisia. Osalle potilaista kehittyy ataksiaa, lihasjäykkyyttä, asento- ja ryhtivirheitä sekä tyypillistä koukistunutta kävelyä. Suurella osalla lapsista todetaan kouluiässä älyllisen kehityksen viivästymä. Aikuisiässä osalle potilaista kehittyy myös liikuntavamma, jonka aste vaihtelee. Kuolleisuus on jonkin verran lisääntynyt.
Etenevät eli progressiiviset myoklonusepilepsiat (PME) ovat monimuotoinen ryhmä harvinaisia keskushermoston rappeutumissairauksia, joiden yhteisiä piirteitä ovat myokloniat, epilepsia ja etenevät neurologiset oireet (8). Peittyvästi periytyvä progressiivinen myoklonusepilepsia tyyppi 1 (EPM1, Unverricht−Lundborgin tauti) on yleisin PME-sairaus maailmassa. Se kuuluu suomalaiseen tautiperintöön, ja sen esiintyvyys Suomessa on 2:100 000 asukasta, eli suurin maailmassa (9).
EPM1 on todennäköisesti alidiagnosoitu, koska sen lievimmät muodot saattavat muistuttaa esimerkiksi nuoruusiän myoklonusepilepsiaa, joka on taustaltaan monitekijäinen. Tauti johtuu tavallisimmin kystatiini B -valkuaisainetta koodaavan CSTB -geenin säätelyalueen toistojaksomutaatiosta.
Ensimmäiset oireet ilmaantuvat yleensä 6-15 vuoden iässä. Ne ovat yleisimmin ulkoisten ärsykkeiden laukaisemia lihasnykäyksiä (myokloniaa) tai tajuttomuus-kouristuskohtauksia (10). Taudin vaikeusaste vaihtelee kliinisiltä oireiltaan ja taudinkulultaan merkittävästi eri perheissä ja myös samassa perheessä.
Diagnoosia seuraavina vuosina myoklonia ja ataksia hankaloituvat, mutta tajuttomuus-kouristuskohtaukset saadaan usein epilepsialääkkeillä hyvin hallintaan. Osalla potilaista liikuntakyky huononee vähitellen niin, että he tarvitsevat pyörätuolia jatkuvasti. Osalla liikunta- ja toimintakyky säilyvät hyvänä läpi elämän. Henkinen suorituskyky ja muisti säilyvät yleensä paremmin kuin liikuntakyky. Ennenaikaista kuolemanriskiä lisäävät 40 ikävuoden jälkeen suurentunut äkkikuoleman vaara, masennus ja siihen liittyvä itsemurhariski, keuhkokuumeen riski ja tapaturmat (9).
Muita PME-sairauksia on tunnistettu useita. Niistä tärkein on lapsuuden neurodegeneratiivinen sairausryhmä neuronaaliset seroidilipofuskinoosit (NCL). Osa NCL-taudeista kuuluu suomalaiseen tautiperintöön (8).
Mitokondriotaudeissa esiintyy monen tyyppisiä epileptisiä kohtauksia. Muissa epilepsioissa myokloonisiin kohtauksiin usein käytettyä valproaattia ei kuitenkaan pidä käyttää maksavaurioriskin vuoksi (11). Mitokondriotaudit voivat johtua joko mitokondrion DNA:n tai tuman genomissa sijaitsevien mitokondriaalisia proteiineja koodittavien geenien mutaatioista. Esimerkiksi mitokondrioiden DNA-polymeraasi gammaa (POLG) koodaavan POLG1 -geenin (12) mutaatioihin liittyvissä taudinkuvissa tyypillisiä piirteitä ovat epilepsia ja hoitoresistentit status epilepticus -tilanteet.
POLG1 -geenin valtamutaatioon liittyy mitokondriaalinen peittyvästi periytyvä ataksia-oireyhtymä (MIRAS). Etenevän pikkuaivoperäisen ataksian ohella sen tyypillisiin piirteisiin kuuluu sensorinen neuropatia, osalla myös vaikea epilepsia. Osalla potilaista ensimmäisenä oireena ovat migreenityyppiset päänsärkykohtaukset ja takaraivolohkoepilepsia.
Moniin kromosomipoikkeavuuksiin ja perimän kopiolukumuutoksiin liittyy merkittävä epilepsiariski. Esimerkiksi rengaskromosomin muodostuessa kromosomin molemmista päistä häviää perimäainesta. Tavallisimmin epilepsian esiintyminen liittyy rengaskromosomeihin 14 ja 20. Rengaskromosomi 20 -oireyhtymä on useimmiten sporadinen, mutta se voi myös periytyä (13). Oireyhtymä on alidiagnosoitu.
Kromosomialueen 15q11q13 mikroduplikaatio -oireyhtymässä henkilöllä on kyseisen alueen trisomia ja puolella esiintyy epilepsiaa. Saman kromosomialueen deleetio puolestaan johtaa joko Prader-Willin tai Angelmannin oireyhtymään siitä riippuen, onko muutos isältä vai äidiltä perityssä kromosomissa (14). Myös näissä oireyhtymissä esiintyy vaikeaa epilepsiaa.
Yöllisestä otsalohkoepilepsiasta noin kolmasosa on vallitsevasti periytyvää alatyyppiä (autosomal dominant nocturnal frontal lobe epilepsy, ADNFLE). Hypermotoriset lyhytkestoiset kohtaukset alkavat unessa, ja ne voidaan diagnosoida virheellisesti unihäiriöiksi. Kuulo-oirein tai afaattisin oirein ilmenevässä ohimolohkoepilepsiassa on todettu myös vallitsevasti periytyvä muoto (autosomal dominant lateral temporal lobe epilepsy, ADLTLE) (15).
Tuberoosiskleroosi on autosomissa vallitsevasti periytyvä neurokutaaninen oireyhtymä, jolle ovat tyypillisiä epilepsia, kehitysvammaisuus ja kasvaimet eri elimissä (16). Epilepsia alkaa usein jo imeväisiässä paikallisalkuisina kohtauksina tai infantiilispasmeina. Noin 70 %:lta potilaista löydetään mutaatio TSC1 - tai TSC2 -kasvunrajoitegeeneissä. Inaktivoiva mutaatio TSC -geeneissä johtaa mTOR-reitin yliaktivoitumiseen, ja tilaa voidaan hoitaa mTOR-estäjillä.
Aivojen kuorikerroksen kehityshäiriöt ovat yksi tärkeimmistä syyryhmistä vaikeassa epilepsiassa. Osa niistäkin aiheutuu mTOR-yliaktivaatioista (15).
Noin 25 % kaikista epilepsioista on geneettisiä yleistyneitä epilepsioita (GGE; aiemmalta nimeltään idiopaattiset yleistyneet epilepsiat), joiden geneettinen tausta on monitekijäinen. Niistä yleisimmät ovat lapsuusiän poissaoloepilepsia, nuoruusiän poissaoloepilepsia ja nuoruusiän myoklonusepilepsia. GGE-sairauksiin liittyviä tärkeitä riskitekijöitä ovat kromosomaaliset deleetiot ja duplikaatiot. GGE-potilaista noin 3 % kantaa yhtä kolmesta toistuvasta deleetiosta kromosomeissa 15 ja 16 (15,17).
Geneettiset tutkimukset kuuluvat osaksi vaikean epilepsian tutkimuksia (taulukko «»1). Ne tehdään ensisijaisesti yliopistosairaalassa tai yliopistosairaalan tai epilepsiaan perehtyneen keskuksen konsultaation perusteella. Selvittelyistä on aina keskusteltava etukäteen potilaan ja perheen kanssa. Potilaalle ja perheelle tulee tarjota mahdollisuus perinnöllisyysneuvontaan geneettisen muutoksen löydyttyä.

Epilepsioiden laaja geenipaneelitutkimus tai eksomisekvensointi kannattaa tehdä potilaille, jotka ovat lapsuudessa sairastuneet etiologialtaan tuntemattomaan epileptiseen enkefalopatiaan tai muuhun vaikeaan epilepsiaan. Tutkimus kannattaa tehdä myös aikuiselle potilaalle.
On kuitenkin muistettava, että toistojaksomuutokset eivät yleensä tule esille geenipaneeleissa tai eksomitutkimuksessa. Molekyylikaryotyypitys on ensisijainen tutkimus potilaille, joilla on epilepsian lisäksi kehityshäiriöitä, älyllinen kehitysvamma tai dysmorfisia piirteitä. Perinteinen kromosomitutkimus on yhä hyvä diagnostiikan väline, jos oirekuvan perusteella on syytä epäillä rengaskromosomioireyhtymää.
Yksittäisten mutaatioiden tutkiminen on perusteltua, jos epilepsian taustalla kliinisen kuvan perusteella ajatellaan olevan ns. valtamutaatio, kuten suomalaisen tautiperinnön sairauksissa. Yksittäisten geenien tarkempi analysointi voi tulla kyseeseen myös potilailla, joilla kliinisten syiden pohjalta epäillään tai löydösten perusteella tiedetään olevan spesifinen sairaus, mutta mutaatiota ei ole löytynyt paneelitutkimuksessa tai eksomisekvensoinnissa.
Suvussa esiintyvän geenivirheen kantajuuden tutkiminen oireettomalta henkilöltä edellyttää aina perinnöllisyyslääkärin antamaa perinnöllisyysneuvontaa ennen tutkimusta. Kantajuutta ei pidä tutkia lapsilta, jos sairaudenkulkua muuttavaa hoitoa ei ole tarjolla eikä lapsi itse pysty antamaan suostumusta.
Epilepsian taustalla olevan geenivirheen löytyminen vahvistaa potilaan diagnoosin ja päättää potilaalle usein raskaan diagnosointiprosessin. Täsmällinen diagnoosi voi antaa tietoa sairauden tyypillisestä kulusta ja ennusteesta. Lisäksi se mahdollistaa potilaan paremman seurannan.
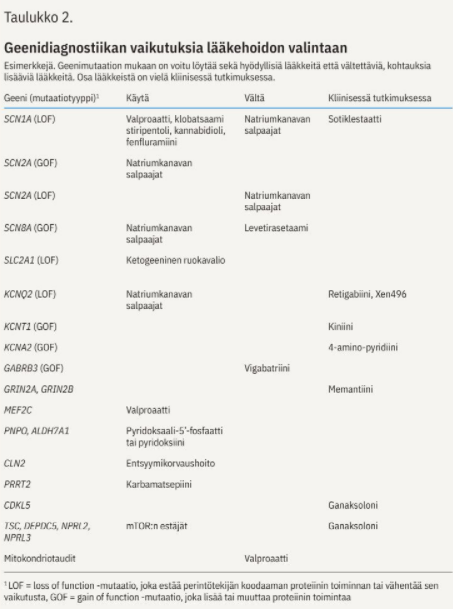
Joissakin tapauksissa geenidiagnoosi vaikuttaa myös epilepsian lääkehoidon valintoihin - jopa saman geenin yksittäisten mutaatioiden erilaisten toiminnallisten ominaisuuksien takia (taulukko «»2) (18,19,20,21). Toistaiseksi geenivirheen löytyminen johtaa silti vain harvoin täsmähoitoon, eli hoitoon, joka kohdistuu epilepsian biologiseen perussyyhyn. Uudet diagnostiset mahdollisuudet, nopeampi päätyminen oikeaan lääkitykseen niiden ansiosta ja turhien tutkimusten välttäminen ovat edullisia kustannusvaikutuksia terveydenhuollolle.
Geenivirheen löytyminen potilaalta mahdollistaa epilepsian perinnöllisyysneuvonnan perheessä. Se tarkentaa merkittävästi arviota taudin uusiutumisriskistä ja antaa perheelle mahdollisuuden harkita alkio- ja sikiödiagnostiikkaa seuraavissa raskauksissa. Kohdennettuja geenitutkimuksia voidaan tarvittaessa tarjota myös lähisukulaisille. On kuitenkin muistettava, että merkittävä osa epilepsiaa aiheuttavista geenimuutoksista on uusia mutaatioita sairastuneella henkilöllä. Lisäksi oirekirjo ja sen vaikea-asteisuus saattavat vaihdella merkittävästi jopa samassa perheessä.
Diagnoosin varmistuminen on yleensä helpotus harvinaista epilepsiaa sairastavalle potilaalle ja hänen perheelleen, vaikka sairauteen ei olekaan parantavaa hoitoa. Se vapauttaa vanhemmat vuosien epätietoisuudesta epilepsian syystä ja mahdollisista itsesyytöksistä (22). Geenidiagnoosin saatuaan potilaat ja perheet voivat hakea vertaistukea.
Koska epilepsia on yleisimmin monitekijäinen sairaus, merkittävä osa epilepsiapotilaista jää yhä ilman geenitason diagnoosia. Perimänlaajuisissa tutkimuksissa tulkintaa vaikeuttaa se, että jokaisen yksilön genomissa esiintyy tuhansia variantteja. Sairautta aiheuttavaa varianttia ei voi aina yksiselitteisesti tunnistaa niiden joukosta. Kun variantin merkitystä ei tunneta, puhutaan ns. VUS-variantista (Variant of Unknown Significance). Niiden merkitystä on syytä tarkastella säännöllisesti uuden tiedon valossa.
Eksomisekvensointi on vähitellen syrjäyttämässä epilepsian geenipaneelitutkimukset. Se saattaa vähitellen syrjäyttää myös molekyylikaryotyypityksen. Kun sekvensointikustannukset pienenevät ja geenitutkimusten kliininen merkitys kasvaa, geenidiagnostiikan tulevaisuuden työkalu lienee koko genomin sekvensointi. Tämä vaatii meiltä jokaiselta varianttitiedon lukutaitoa.