

Ks. artikkelin pdf-versio «http://www.fimnet.fi/cl/laakarilehti/pdf/2023/SLL13-14-2023-543.pdf»1 Lääkärilehden sivuilla (vaatii FiMnet-tunnuksen).
Rintasyöpä on naisten yleisin syöpä. Siihen sairastuu vuosittain maailmassa yli 2 000 000 naista ja Suomessa yli 5 000, elinikäisen riskin ollessa 13 % (1). Vuonna 2019 rintasyöpään menehtyi noin 900 naista Suomessa (2,3).
Rintasyövän on jo kauan tiedetty esiintyvän perheittäin. Kansainvälinen tiedeyhteisö on aktiivisesti tutkinut siihen liittyvää perinnöllistä alttiutta, jotta osa syövistä voitaisiin ehkäistä tai todeta mahdollisimman varhain.
Ensimmäiset suureen rintasyöpäriskiin liittyvät ja edelleen tärkeimmät geenit BRCA1 ja BRCA2 tunnistettiin jo 1990-luvun puolessavälissä (4,5), jolloin näiden geenien aktiivinen tutkiminen alkoi kliinisessä työssä suomalaisissa suuren riskin perheissä. Sittemmin on tunnistettu riskiryhmiä, joille geneettiset tutkimukset ovat aiheellisia sukutaustasta riippumatta.
Epäily rintasyövän perinnöllisyydestä on yleisin perinnöllisyyslääketieteellisiin tutkimuksiin lähettämisen syy maailmassa. Tutkittaessa sukuja havaittiin pian, että kliinisessä työssä rintasyöpäriski voidaan jakaa kolmeen ryhmään: suuri riski (yli 40 %:n elinikäinen riski), kohtalainen riski (20-40 %:n elinikäinen riski) ja vähäinen riski, joka ei merkittävästi eroa väestöriskistä (alle 20 %:n elinikäinen riski) (6). Tämä jako vahvistui, kun geenejä ja variantteja on tunnistettu jokaiseen ryhmään (7).
Suuren riskin alttiuteen viittaavat useiden rintasyöpätapausten esiintyminen suvussa, rintasyöpädiagnoosi alle 40 vuoden iässä, medullaarinen tai kolmoisnegatiivinen rintasyöpä alle 60-vuotiaana, munasarjasyövän esiintyminen lähisuvussa rintasyövän kanssa ja rintasyöpä miehellä (6). Geenitutkimusten siirryttyä BRCA1/2-geeneistä useiden geenien tutkimukseen geenipaneeleilla, suuren riskin ja useat kohtalaisen riskin geenit tulevat tutkituiksi samanaikaisesti.
Tilanteissa, jotka eivät edellytä sukutaustan selvittämistä, geenitutkimuksia tehdään useilla erikoisaloilla. Jos geenivirhe todetaan tai suvun selvittely on aiheellista, potilas tulee lähettää perinnöllisyyslääketieteen yksikön konsultaatioon ja jatkotutkimuksiin.
Suuren rintasyöpäriskin geenejä ovat BRCA1, BRCA2, PALB2, PTEN, TP53, STK11 ja CDH1 (taulukko «»1).
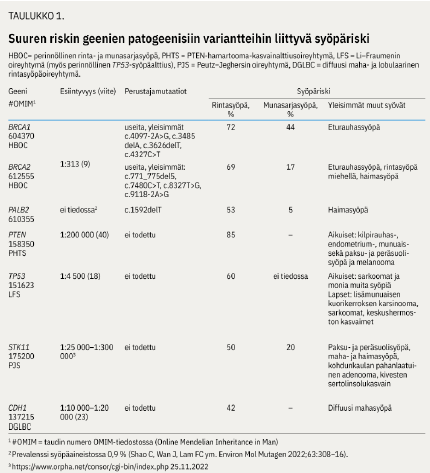
BRCA1- ja BRCA2-geenit ovat kasvunrajoitegeenejä, jotka osallistuvat DNA:n vaurioiden korjaamiseen homologisen rekombinaation mekanismeilla (8). BRCA1-geenivirheiden esiintyvyys väestössä on noin 0,2 % ja BRCA2-virheiden noin 0,3 % (9).
Väestöön rikastuneet ns. perustajamutaatiot kattavat Suomessa noin 84 % kaikista BRCA1/2-geenivirheistä, mutta esiintyvyydessä on alueellisia eroja (10-12). Suuressa aineistossa BRCA1-geenivirhettä kantavien naisten elinikäiseksi rintasyöpäriskiksi arvioitiin 72 % ja munasarjasyövän riskiksi 44 %, BRCA2-geenivirhettä kantavilla naisilla riskit olivat vastaavasti 69 % ja 17 % (13). BRCA2-geenivirheisiin näyttää liittyvän suurentunut rintasyöpäriski myös miehillä sekä 2-5-kertainen eturauhassyövän riski, BRCA1-varianttien kantajilla riskit ovat pienempiä (14).
PALB2 on kasvunrajoitegeeni, joka osallistuu DNA:n vaurioiden korjaukseen, ja siten sen patogeeniset (PV) ja todennäköisesti patogeeniset variantit (LPV) aiheuttavat lisääntyneen syöpäriskin (15). PALB2-geeniä pidetään kolmantena tärkeänä rintasyövälle altistavana geeninä BRCA1- ja BRCA2-geenien ohella. PALB2-geenin elinikäinen rintasyöpäriski asettuu suuren riskin ryhmään (53 %) (16). Suvun syöpähistoria on kuitenkin merkityksellinen arvioitaessa PALB2-geenivirheeseen liittyvää riskiä. Kansainvälisten suositusten mukaisesti rintojen seuranta olisi sama kuin BRCA-geenivirheen kantajilla. PALB2-patogeenisiin variantteihin liittyy suurentunut riski myös munasarjasyöpään (5 %), haimasyöpään (2-3 %) ja miesten rintasyöpään (1 %). Haimasyövän suhteen sukuhistoria vaikuttaa siihen, tarvitaanko seurantaa (16).
PTEN-hamartooma-kasvainalttiusoireyhtymä (PHTS) kuvattiin 1990-luvun lopulla. PTEN on kasvunrajoitegeeni, fosfataasi- ja tensiinihomologi. Oireyhtymä ilmenee useiden kudosten lukuisina hamartoomina ja lisääntyneenä riskinä sairastua erityisesti rintasyöpään; elinikäinen riski on jopa 85 %. Lisäksi kilpirauhasen syövän riski on suurentunut (35 %), samoin kuin kohdun limakalvon syövän (28 %), munuaissyövän (34 %), paksu- ja peräsuolen syöpien (20 %), melanooman (6 %) ja erittäin harvinaisen aivokasvaimen, Lhermitte-Duclos'n taudin. PTEN-geenin haitalliset muutokset ovat yhteydessä Cowdenin ja Bannayan-Riley-Ruvalcabanin oireyhtymiin sekä Proteus-oireyhtymää muistuttavaan oireyhtymään (17).
TP53-geenin ituratamuutokset aiheuttavat Li-Fraumenin syöpäoireyhtymän. TP53 reagoi DNA-vaurioihin säätelemällä monien geenien ilmenemistä ja siten mm. DNA:n korjausmekanismeja, apoptoosia ja angiogeneesiä. TP53-ituratamuutos todetaan usein syöpään sairastuneella lapsella tai nuorella rintasyöpäpotilaalla, jolla ei ole syöpää suvussa (18). Negatiivinen sukuhistoria ei siis sulje pois TP53-ituratamuutosta, sillä niistä 7-20 % on de novo -muutoksia. Näistä noin 20 % tapahtuu sikiökaudella, ja siksi ne ovat mosaiikkisia ja penetranssi on epätäydellinen. Syöpäriskit arvioitiin aiemmin perheiden perusteella, mutta sittemmin kliinisen kuvan on todettu olevan hyvin vaihteleva mm. patogeenisen variantin sijainnin ja proteiinirakenteen muutoksen mukaan. TP53-variantteihin liittyy hyvin monien syöpien riski (taulukko «»1). Myös munasarjasyövän riski näyttäisi olevan jonkin verran lisääntynyt (19).
STK11-geenin patogeeniset muutokset liitetään vallitsevasti periytyvään Peutz-Jeghersin syöpäalttiusoireyhtymään (PJS). STK11-geenin tuote säätelee mm. solujen metaboliaa sekä proliferaatiota toimien kasvunrajoitegeeninä. Oireyhtymä on polypoosi, johon liittyy suurentunut ruoansulatuskanavan hamartomatoottisten polyyppien riski. Maha-suolikanavan syöpien kumulatiivinen riski on 10-40 %, ja suurin riski liittyy paksu- ja peräsuolisyöpään. Oireyhtymässä esiintyy myös muita syöpiä, joista merkittävimpinä naisten rintasyöpä (30-50 %), haimasyöpä (10-35 %) sekä kivesten sertolinsolukasvaimet (n. 10 %). Erilaisten munasarjakasvainten, etenkin SCTAT-kasvainten (sex-cord tumor with annular tubules), riski on noin 20 %. Geenimuutoksiin, jotka aiheuttavat lyhentyneen proteiinituotteen muodostumisen (ns. trunkoivat mutaatiot), liittyy yleensä vaikeampi taudinkuva (20).
CDH1-geenin tuottama epiteliaalinen kadheriini (E-kadheriini) toimii solujen välisen adheesion muodostumisessa, epiteelisolujen erilaistumisessa ja polarisaatiossa. CDH1:n ituradan patogeenisten varianttien ja perinnöllisen diffuusin mahasyövän (hereditary diffuse gastric cancer, HDGC) yhteys kuvattiin 1998 (21). Sittemmin on todettu, että CDH1-geenin patogeeniset variantit lisäävät myös naisen rintasyöpäriskiä (21). CDH1-geenin patogeenisiä inaktivoivia variantteja tunnetaan kymmenittäin. Tyypillisesti ne aiheuttavat lyhentyneen proteiinin, mutta myös patogeenisiä missense-muutoksia tunnetaan. Patogeenistä varianttia kantavilla miehillä diffuusin mahasyövän kumulatiivinen riski 80 ikävuoteen mennessä on 70 % ja naisilla 56 %, ja lobulaarisen rintasyövän elinikäinen riski on 42 % (22,23). Suun ja kasvojen alueen halkiot voivat joskus liittyä CDH1-geenin variantteihin joko syöpäriskin kanssa tai ilman sitä.
Suomessa todettiin vuosina 2015-19 yhteensä 147 rintasyöpää miehellä, eli vuosittain tapauksia on noin 30 (2). Miehillä perinnöllinen alttius rintasyöpään liittyy ainakin BRCA1-, BRCA2- ja PALB2-variantteihin, joissa rintasyöpäriski on vastaavasti 0-4 %, 4-16 % ja 1 % (16,24,25).
Joissakin geenipaneelitutkimuksissa positiivisten löydösten osuus miesten rintasyövässä on ollut lähes 20 % (26). Positiivisten osuuteen vaikuttaa BRCA2-patogeenisten varianttien yleisyys väestössä. Suomalaisessa tutkimuksessa BRCA2-varianttien esiintyvyys koko aineistossa oli 7,8 %. Lisäksi miehillä, joilla oli rinta- tai munasarjasyöpää suvussa, esiintyvyys oli 44 %, vaikka osalta oli tutkittu vain kahdeksan suomalaisen valtamutaation esiintyvyys (27).
Miehille ei Suomessa ole järjestetty säännöllistä seurantaa eikä kuvantamistutkimuksia rintasyöpäriskin vuoksi. Geenivirheen kantajien tunnistaminen on kuitenkin tärkeää, jotta heille voidaan kertoa syöpäriskistä ja antaa ohjeet omatoimisesta rintarauhaskudoksen tutkimisesta ja hakeutumisesta hoitoon tarvittaessa.
Syöpäriskin arviointi perustuu tilastolliseen riskiin, eikä kliinisessä työssä ole toistaiseksi arvioitu syöpäriskiä henkilökohtaisesti. Tiedetään kuitenkin, että tiettyjen alueiden variantit esimerkiksi BRCA-geeneissä altistavat enemmän rinta- tai munasarjasyövälle (28) ja tietyissä geeneissä variantin vaikutus proteiinirakenteeseen määrittää oirekuvaa (20). Henkilökohtaiseen syöpäriskiin vaikuttavat monet seikat, kuten ympäristötekijät, genomin satunnaiset somaattiset variantit, mutta myös muut ituradan variantit, joita tutkimalla voitaisiin tarkentaa henkilökohtaista riskiarviota (29).
Henkilökohtaisen riskin arvioinnilla olisi erityisen suuri merkitys pohdittaessa riskiä vähentäviä toimenpiteitä vaihtoehtona seurannalle. Nyt seuranta on aktiivisesti geenitutkimuksia tekevissä maissa hyvin yhdenmukaista NCCN:n (National Comprehensive Cancer Network) ja NICE:n (National Institute of Health and Care Excellence) julkaisemiin yleisiin suosituksiin sekä ERN-Genturiksen geenikohtaisiin suosituksiin perustuvaa (30-32).
Rintasyöpäalttiutta seurataan kuvantamistutkimuksin: rintojen magneettitutkimuksella, mammografialla ja vähäisemmässä määrin kaikututkimuksella (taulukko «»2). Periaatteena on tehdä magneettikuvaus vuosittain 50-60 vuoden ikään saakka ja siirtyä sen jälkeen mammografiaan. Yli 70-vuotiaille suositellaan mammografiaa kahden vuoden välein, yleistila huomioon ottaen. Munasarjasyövän riskin vuoksi ei Suomessa yleensä suositella seurantaa vaan riskiä vähentävää toimenpidettä, joka ratkaistaan geenikohtaisesti iän mukaan (33).

Vaihtoehtona seurannalle ovat riskiä vähentävä mastektomia (RRM) ja rekonstruktio sekä riskiä vähentävä munasarjojen ja munanjohdinten poisto (RRSO). Sekä terveen kantajan bilateraalisen että rintasyöpään sairastuneen kontralateraalisen rinnan poiston on todettu vähentävän rintasyövän esiintyvyyttä familiaalisessa riskissä ja BRCA-geenivirheiden kantajilla (34-36). Samoin munasarjojen ja munanjohdinten poisto vähentää merkittävästi munasarjasyövän riskiä BRCA1- ja BRCA2-geenivirheen kantajilla (36,37). Molempien toimenpiteiden jälkeen jäännösriski on alle 5 %. Suomessa riskiä vähentäviä mastektomioita on tutkittu Hyksin potilailla, joista 38 %:lle oli tehty bi- tai kontralateraalinen rinnanpoisto (38). Suositukset toimenpiteiden ajankohdasta vaihtelevat geenikohtaisesti (taulukko «»2).
Suuren riskin patogeenisten varianttien kantajien hoito eroaa osittain muiden rinta- ja munasarjasyöpäpotilaiden hoidosta. Heille ei yleensä tehdä säästäviä rintasyöpäleikkauksia, mikäli kantajuus on tiedossa toimenpiteen ajankohtana. Munasarjasyövän ja rintasyövän hoidossa voidaan käyttää poly-ADP-riboosipolymeraasin (PARP) estäjiä. Soluissa, joissa on DNA:n kaksoisjuosteita korjaavan homologisen rekombinaation toimintahäiriö, PARP:n esto johtaa solujen kuolemaan (39).
Rintasyöpään liittyvää perinnöllistä alttiutta on tutkittu jo yli 30 vuotta, mutta BRCA1/2-geenit ovat edelleen yleisimmät suuren riskin alttiusgeenit, vaikka uusiakin geenejä on tunnistettu. Näiden geenien rinnalle geenipaneeleihin ovat tulleet kohtalaisen riskin geenit.
Suuren riskin seurantalinjat ovat melko yhdenmukaisia, mutta seurannassa ja riskiä vähentävien toimenpiteiden perustelussa ja ajankohdassa on geenikohtaisia eroja. Tavoitteemme on, että toimintalinjat olisivat yhdenmukaiset koko Suomessa.
Lue myös alkuperäistutkimus s. 548